Introduction
Acute lung injury (ALI), a common complication of sepsis, is characterized by lung inflammation, impaired gas exchange, and respiratory distress [1]. Recent data indicate that sepsis accounts for approximately 10 million global deaths annually, with sepsis-induced ALI exhibiting a mortality rate of 30-40% [2]. The pathogenesis of ALI involves multifactorial mechanisms, including inflammation, oxidative stress, and regulated death processes such as pyroptosis. Despite therapeutic advances, in-hospital mortality rates for ALI remain alarmingly high (38-46%) [3]. Notably, a growing body of evidence underscores pyroptosis, a lytic inflammatory form of cell death, as a critical driver of ALI progression [4]. Therefore, understanding the molecular mechanism of pyroptosis in ALI is essential for developing targeted therapies to mitigate lung injury and improve clinical outcomes.
Pyroptosis can be triggered in lung epithelial cells and endothelial cells as part of the immune response to pathogens [5]. A previous study demonstrated that pyroptosis inhibition reduced inflammatory cascades, pulmonary edema, and lung injury in sepsis-induced ALI models [6]. Nucleotide-binding oligomerization domain (NOD)-like receptor family, pyrin domain containing 3 (NLRP3) plays a key role in the initiation and execution of pyroptosis in ALI through the formation of inflammasome and the release of caspase-1 and inflammatory cytokines. For example, puerarin alleviated ALI through suppressing pyroptosis by reducing NLRP3 inflammasome assembly [7]. Similarly, the inhibition of C3a/C3a receptor (C3aR) axis repressed endothelial pyroptosis, thereby improving sepsis-induced ALI outcomes [8]. Another study showed that treatment with double-stranded RNA-dependent kinase (PKR) significantly ameliorated lipopolysaccharide (LPS)-induced ALI in mice through suppressing NLRP3-mediated pyroptosis [9]. However, the precise regulatory mechanisms of NLRP3-driven pyroptosis in ALI remain unclear.
Deltex E3 ubiquitin ligase 3-like (DTX3L), a member of the Deltex protein family, regulates ubiquitination-dependent processes and modulates immune signaling [10]. While DTX3L is implicated in diverse pathologies, such as enhancing epidermal growth factor receptor (EGFR) ubiquitination to drive pancreatic tumorigenesis [11], its role in ALI is unexplored. In antiviral immunity, DTX3L interacts with histone H2B family member J (H2BJ) to amplify interferon responses [12]. Moreover, DTX3L knockdown activated the cGAS/STING pathway via cGAS ubiquitination, thus triggering the production of pro-inflammatory cytokines in pancreatic cancer [13]. Intriguingly, a study demonstrated that DTX3L suppressed pyroptosis in oxygen-glucose deprivation/reperfusion (OGD/R)-induced retinal cells by promoting NLRP3 ubiquitination [14]. Whether DTX3L similarly regulates NLRP3 ubiquitination to modulate pyroptosis in sepsis-induced ALI warrants investigation.
Krüppel-like factor 14 (KLF14), a transcriptional regulator of inflammatory pathways, has emerged as a pivotal regulator in inflammatory diseases [15]. As reported, KLF14 upregulation mitigated neuroinflammation in ischemic-reperfusion injury, suggesting its potential in modulating inflammatory responses [16]. Hu et al. revealed that KLF14 effectively repressed the inflammatory cascade in endothelial cells by inhibiting kappa-light-chain-enhancer of activated B cells (NF-κB) signaling [17]. Notably, KLF14 overexpression protected alveolar epithelial cells from death, thereby attenuating LPS-induced ALI [18]. JASPAR, an open-access resource of transcription factor binding profiles, predicts that KLF14 can bind to the promotor region of DTX3L, suggesting transcriptional regulation. Nevertheless, the functional relationship between KLF14, DTX3L, and pyroptosis in ALI remains uncharacterized.
Based on these findings, we hypothesized that KLF14 suppressed NLRP3-mediated pyroptosis by transcriptionally activating DTX3L, thereby ameliorating sepsis-induced ALI. This study indicates that targeting the KLF14-DTX3L-NLRP3 axis may be a potential therapeutic strategy for sepsis-induced ALI management.
Material and methods
Cell culture and treatment
BEAS-2B, a human bronchial epithelial cell line, responds predictably to cytokines, oxidative stress, and other stimuli typically associated with respiratory tract exposure to environmental agents. Hence, it is widely used as an in vitro cell model for studying respiratory diseases, including ALI [19]. BEAS-2B cells were obtained from American Type Culture Collection (ATCC, Virginia, USA) and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Sigma-Aldrich, Missouri, USA) supplemented with 10% fetal bovine serum (FBS) and 100 µg/ml penicillin-streptomycin (Thermo Scientific, Massachusetts, USA) at 37°C under 5% CO2. To establish a sepsis-induced ALI model, cells were treated with 1 µg/ml LPS (Thermo Scientific) for 12 h after 24 h of initial culture.
Cell transfection and construction of plasmid
For construction of KLF14 or DTX3L overexpression plasmid, full-length KLF4 and DTX3L sequences were cloned and inserted into the pcDNA3.1 vector. For knockdown, short hairpin RNA (sh-RNA) targeting DTX3L (sh-DTX3L) was synthesized. These plasmids and negative control (NC) plasmids were synthesized by GenePharma (Shanghai, China). BEAS-2B cells were transfected with plasmids or shRNA using Lipo6000 reagent (Beyotime, shanghai, China) and incubated for 48 h.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen, California, USA). Then, 1 µg RNA was reverse transcribed into complementary DNA (cDNA) using a Prime-Script one-step qRT-PCR kit (TAKARA, Kyoto, Japan). Amplification was performed on the ABI 7500 Fast RealTime PCR System (Thermo Scientific) with the following primers: KLF14 (NM_138693.4) forward 5’-AAAAGTGGGGGTTGCCTCAA-3’, reverse 5’-GTGGATGGGTGAGACACCAG-3’; DTX3L (NM_138287.3) forward 5’-CTTCCGGGTGGAGTTCAGTG-3’, reverse 5’- TGGGCACAGGTTTTTCGTCA-3’; TRIM24 (NM_003852.4) forward 5’-ATATGCAGCAACAGCAACCG-3’, reverse 5’-GGGCTGGAAGGAGTAGAGGA-3’; TRIM65 (NM_001256124.2) forward 5’-CGCCAACCGTCACTTCTATCT-3’, reverse 5’-ACAGGGTCAGGGTCCTACC-3’; MARCH7 (NM_001282805.2) forward 5’-TCACAGTCCCGTAGTAATGTACC-3’, reverse 5’-CGCCTAAGAAATCGAAATCCCTG-3’; RBX1 (NM_014248.4) forward 5’-TTGTGGTTGATAACTGTGCCAT-3’, reverse 5’-GACGCCTGGTTAGCTTGACAT-3’; GAPDH (NM_001256799.3) forward 5’-ACAACTTTGGTATCGTGGAAGG-3’, reverse 5’-GCCATCACGCCACAGTTTC-3’. All primers were synthesized by Tsingke (Beijing, China). Relative mRNA expression was calculated using the 2-ΔΔCT method with GAPDH serving as the internal control.
Western blot
Proteins were extracted from BEAS-2B cells using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime) and quantified via BCA assay (Beyotime). Proteins were separated via 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (Thermo Scientific) and transferred to polyvinylidene fluoride (PVDF) membranes (Thermo Scientific). After washing with tris-buffered saline with Tween (TBST), the membranes were blocked with non-fat milk and then incubated overnight at 4°C with the following antibodies: Anti-NLRP3 antibody (ab283819, 1 : 1000, Abcam, Cambridge, USA), Anti-Apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) antibody (ab309497, 1 : 1000, Abcam), anti-caspase-1 antibody (ab62698, 1:500), Anti-Gasdermin D (GSDMD) antibody (ab210070, 1 : 1000, Abcam), Anti-tripartite motif-containing protein 24 (TRIM24) antibody (ab256491, 1 : 1000, Abcam), Anti-tripartite motif-containing protein 65 (TRIM65) antibody (ab154821, 1 : 1000, Abcam), Anti-RING-box protein 1 (RBX1) antibody (ab133565, 1 : 5000, Abcam), Anti-membrane-associated RING-CH E3 ubiquitin ligase 7 (MARCH7) antibody (#MBS8530016, 1 : 500, Mybiosource, California, USA), Anti-DTX3L antibody (#MBS6004966, 1 : 1000, Mybiosource). Following washing, membranes were then incubated with Goat Anti-Rabbit IgG H&L preadsorbed (ab7085, 1: 500, Abcam), for 45 min at room temperature. Protein bands were visualized and analyzed using a Tanon-4500 digital image processing system (Tanon, Shanghai, China) and Image-Pro Plus software (Media Cybernetics, Inc., Maryland, USA).
Cell Counting Kit-8 (CCK-8)
BEAS-2B cells (1.0 × 104) were planted into 96-well plates and incubated overnight. Afterwards, the cells were supplemented with 10 µl of CCK-8 solution (Sigma-Aldrich), followed by further incubation at 37°C for another one hour. The absorbance of the solutions at 450 nm was recorded using a scanning microplate reader (Bio-Rad, California, USA).
Flow cytometry
After rinsing, BEAS-2B cells were re-suspended in 50 µl of phosphate-buffered saline (PBS, Thermo Scientific) containing 1% FBS. The samples were then incubated with Alexa Fluor 488-conjugated caspase-1 FLICA (Bio-Rad) at 37°C for 1 h. After centrifugation and washing, the samples were stained with PI staining buffer. Finally, the stained cells were examined using a flow cytometer (BD Biosciences, New Jersey, USA), with pyroptosis identified as double positivity for FAM-YVAD-FMK and PI staining.
Enzyme-linked immunosorbent assay (ELISA) and lactate dehydrogenase (LDH) activity
The levels of interleukin (IL)-18 and IL-1β in cell supernatants were quantified using ELISA assay with commercial kits (#MBS2701000 and #MBS3803011, Mybiosource). Cell culture supernatants were collected and stored at –80°C. The optical density (OD) values were measured at 450 nm using a microplate reader (Thermo Scientific), and IL-18 and IL-1 β levels were calculated. LDH release was assessed using a LDH kit (#MBS9718969, Mybiosource).
Chromatin immunoprecipitation (ChIP)
BEAS-2B cells were crosslinked with 1% formaldehyde (Thermo Scientific) at 37°C for 10 min. After cell lysis, nuclei were collected and resuspended in lysis buffer (Applied Biosystems, California, USA), followed by sonication at 4°C to fragment the DNA. The sonicated lysates were then incubated overnight with beads conjugated with KLF14 antibody (LS-C101601, LifeSpan BioSciences, Inc., Washington, USA). The obtained DNA-protein complexes were washed and exposed to ribonuclease A (RNase A) and proteinase K to isolate purified DNA. The enrichment of DTX3L promoter regions in the KLF14-bound fraction was identified using quantitative polymerase chain reaction (qPCR).
Dual-luciferase reporter assay
JASPAR database predicted three binding sites for KLF14 on the DTX3L promoter. To validate their interaction, DTX3L promoter fragments containing predicted KLF14 binding sites were cloned into the luciferase reporter vector pGL4.10 (Promega Corporation, Wisconsin, USA). BEAS-2B cells were co-transfected with KLF14 overexpressing plasmid along with 100 ng of reporter vectors using Lipofectamine 3000 (Invitrogen). Luciferase activity was calculated after 48 h using a dual-luciferase reporter system (Promega Corporation).
Co-immunoprecipitation (Co-IP)
BEAS-2B cells were lysed with immunoprecipitation (IP) lysis buffer (Meilunbio, Nanjing, China) containing a protease inhibitor cocktail for 30 min. The cell lysates were centrifuged at 4°C for 5 min. Subsequently, cell lysates were incubated overnight 4°C with Protein A/G-Agarose Beads (Invitrogen,) conjugated with anti-NLRP3 antibody (ab263899, Abcam). After washing four times with PBS, the bead-protein complexes were boiled at 100°C for 10 min to elute the proteins, which were subsequently analyzed by western blot.
Statistical analysis
The data were expressed as mean ± standard deviation (SD). Comparison between two groups was performed using Student’s t test. Otherwise, multiple groups were analyzed via one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Statistical significance was set at p < 0.05. GraphPad prism 8 software (GraphPad, San Diego, California, USA) was used for data analysis.
Results
Upregulation of KLF14 inhibited NLRP3-mediated pyroptosis in LPS-induced BEAS-2B cells
To investigate the impact of KLF14 on pyroptosis, BEAS-2B cells were treated with LPS. First, KLF14 overexpression efficiency was validated, showing a significant increase in its expression (Fig. 1A). Compared to the control group, KLF14 expression was downregulated in LPS-treated BEAS-2B cells, while its expression was enhanced upon KLF14 overexpression (Fig. 1B). LPS treatment reduced cell viability and exacerbated pyroptosis, whereas KLF14 overexpression counteracted these effects (Fig. 1C, D). Consistent with these findings, LPS exposure upregulated the levels of NLRP3, ASC, caspase-1 and GSDMD, which were reversed by KLF14 overexpression (Fig. 1E). Moreover, KLF14 upregulation reduced the secretion of IL-18 and IL-1β in LPS-stimulated BEAS-2B (Fig. 1F). Furthermore, LPS-induced activation of extracellular LDH activity was suppressed by KLF14 overexpression (Fig. 1G). Collectively, these findings demonstrated that increased KLF14 expression exerted inhibitory effects on NLRP3-mediated pyroptosis in LPS-induced BEAS-2B cells.
Fig. 1
Upregulation of KLF14 inhibited NLRP3-mediated pyroptosis in LPS-induced BEAS-2B cells. A) BEAS-2B cells were exposed to 1 μg/ml LPS. KLF14 mRNA was detected using qRT-PCR. OE-KLF14 or OE-NC was transfected into BEAS-2B cells treated with LPS. B) KLF14 mRNA was detected using qRT-PCR analysis. C) Cell viability was assessed using CCK-8 assay. D) Pyroptosis rate was determined using flow cytometry. E) Protein levels of NLRP3, ASC, caspase-1, and GSDMD were quantified by western blot.F) The contents of IL-18 and IL-1β were evaluated by ELISA kits. G) The release of LDH was detected by a commercial kit. Data are shown as mean ±SD. *p < 0.05, **p < 0.01, ***p < 0.001. KLF14 – Krüppel-like factor 14, LPS – lipopolysaccharide, OE-KLF14 – overexpression of KLF14, OE-NC – overexpression negative control, qRT-PCR – quantitative reverse transcription polymerase chain reaction, CCK-8 – Cell Counting Kit-8, ASC – apoptosis-associated speck-like protein containing a CARD, GSDMD – gasdermin D, IL-18 – interleukin 18, IL-1β – interleukin 1β, LDH – lactate dehydrogenase
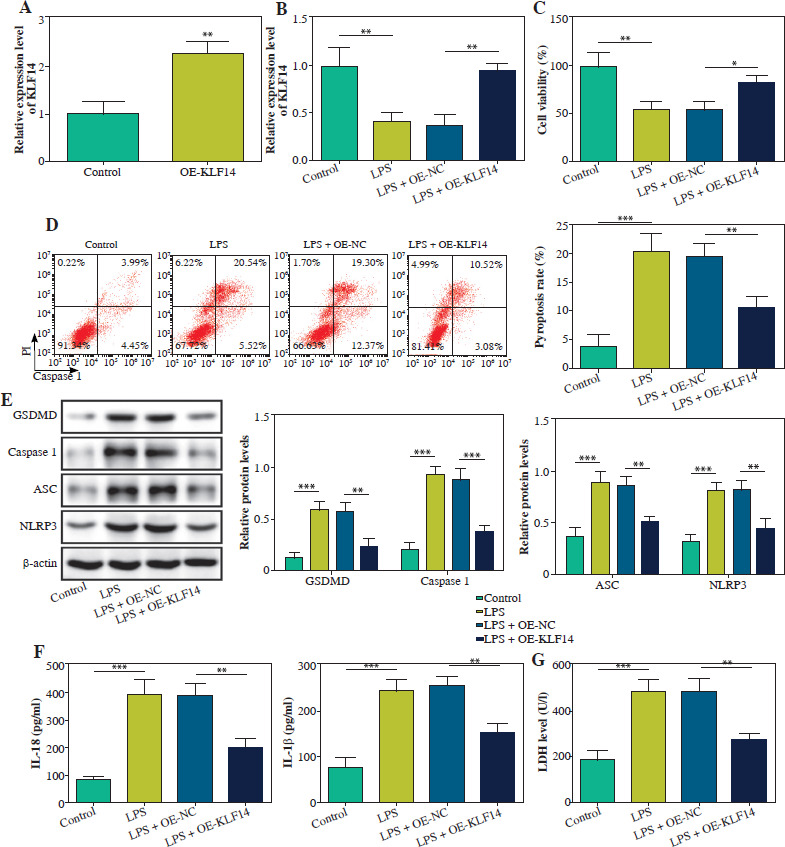
KLF14 transcription activated DTX3L expression in BEAS-2B cells
We screened five E3 ubiquitin ligases (TRIM24, DTX3L, TRIM65, MARCH7, RBX1) known to promote NLRP3 ubiquitination, and examined their transcriptional regulation by KLF14. As displayed in Figure 2A, B, KLF14 overexpression remarkably increased DTX3L expression but had no effect on TRIM24, TRIM65, MARCH7, and RBX1. JASPAR database analysis predicted three potential KLF14 binding sites in the DTX3L promoter region (Fig. 2C). The ChIP analysis confirmed that KLF14 predominantly bound to site 1 of the DTX3L promoter, with minimal enrichment at sites 2 and 3 (Fig. 2D). Luciferase reporter assays further demonstrated that KLF14 overexpression enhanced luciferase activity driven by Pro or Pro#1, but had no effect on Pro#2 and Pro#3 (Fig. 2E). These results illustrated that KLF14 transcriptionally activated DTX3L by directly binding to its promoter.
Fig. 2
KLF14 transcription activated DTX3L expression in BEAS-2B cells. A) OE-KLF14 or OE-NC was transfected into BEAS-2B cells. mRNA expression levels of TRIM24, DTX3L, TRIM65, MARCH7 and RBX1 were calculated by qRT-PCR. B) Protein levels of TRIM24, DTX3L, TRIM65, MARCH7 and RBX1 were detected using western blot. C) The binding site between KLF14 and DTX3L was predicted by JASPAR. D) The interaction between KLF14 and DTX3L was identified by ChIP assay. E) The regulatory effect of KLF14 on DTX3L was verified by dual-luciferase reporter assay. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. DTX3L – Deltex E3 ubiquitin ligase 3 like, OE-KLF14 – overexpression of KLF14, OE-NC – overexpression negative control, qRT-PCR – quantitative reverse transcription polymerase chain reaction, TRIM24 – tripartite motif containing 24, TRIM65 – tripartite motif containing 65, MARCH7 – membrane-associated ring-ch 7, RBX1 – RING Box 1, ChIP – chromatin immunoprecipitation
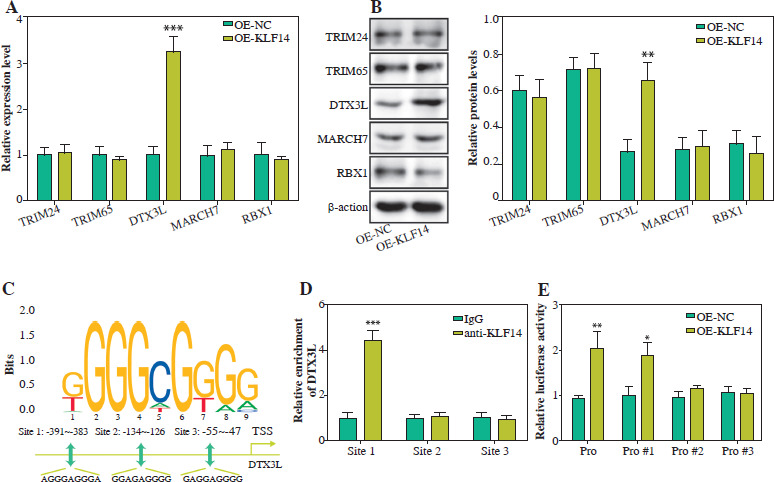
DTX3L degraded NLRP3 through ubiquitinating NLRP3 in BEAS-2B cells
To elucidate the downstream mechanism of DTX3L in ALI, we generated stable DTX3L-overexpressing BEAS- 2B cells (Fig. 3A). To inhibit proteasomal or lysosomal activity, we used MG132 and chloroquine (CQ) to treat cells. The results showed that elevated DTX3L markedly reduced NLRP3 protein levels, with MG132 treatment reversing the inhibitory effect of DTX3L, and CQ treatment exhibited no effect on it (Fig. 3B). Co-IP assay confirmed that DTX3L was significantly precipitated in the anti- NLRP3 group compared to the control group, suggesting the interaction between DTX3L and NLRP3 (Fig. 3C), and an increase in DTX3L was found to enhance the ubiquitination of NLRP3 (Fig. 3D). Next, BEAS-2B cells were exposed to cycloheximide (CHX) for 0, 3, 6, 9 and 12 h. It revealed that increased DTX3L led to decreased NLRP3 protein stability and accelerated NLRP3 protein degradation (Fig. 3E). In conclusion, DTX3L degraded NLRP3 protein by promoting its ubiquitination in BEAS-2B cells.
Fig. 3
DTX3L degraded NLRP3 through ubiquitinating NLRP3 in BEAS-2B cells. A) OE-DTX3L or OE-NC was transfected into BEAS-2B cells. DTX3L mRNA was quantified using qRT-PCR. B) Cells were transfected with OE-DTX3L, OE-NC, OE-DTX3L with MG132 or CQ. NLRP3 protein was evaluated using western blot. C) The interaction between DTX3L and NLRP3 was validated by co-IP. D) Ubiquitination level of NLRP3 was assessed using western blot. E) DTX3L overexpressing BEAS-2B cells were exposed to CHX for 0, 3, 6, 9, and 12 h. Protein stability of NLRP3 was assessed using western blot. Data are shown as mean ±SD. *p < 0.05, **p < 0.01, ***p < 0.001. DTX3L – Deltex E3 ubiquitin ligase 3 like, OE-DTX3L – overexpression of DTX3L, OE-NC – overexpression negative control, qRT-PCR – quantitative reverse transcription polymerase chain reaction, MG132 – A proteasome inhibitor, CQ – chloroquine, Co-IP – co-immunoprecipitation, CHX – cycloheximide
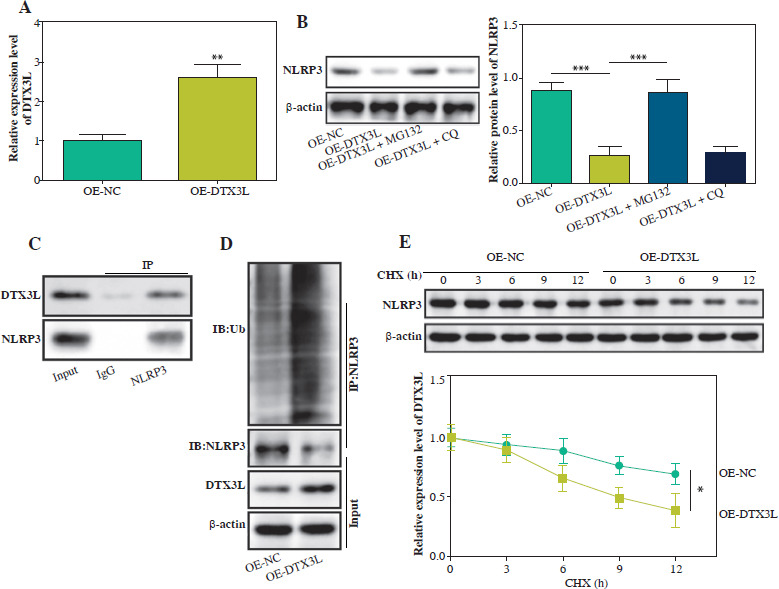
DTX3L suppressed NLRP3-mediated pyroptosis in LPS-induced BEAS-2B cells
We further explored the functional role of DTX3L in BEAS-2B cells. As depicted in Figure 4A, B, LPS treatment repressed cell viability and enhanced pyroptosis, whereas DTX3L overexpression reversed these effects. It also reversed LPS-induced upregulation of NLRP3, ASC, caspase-1 and GSDMD (Fig. 4C). Furthermore, the LPS-triggered increase in IL-18 and IL-1 β was reduced by DTX3L overexpression (Fig. 4D). The elevated extracellular LDH release induced by LPS was also repressed by upregulating DTX3L (Fig. 4E). These data proved that DTX3L repressed NLRP3-mediated pyroptosis in LPS-stimulated BEAS-2B cells.
Fig. 4
DTX3L suppressed NLRP3-mediated pyroptosis in LPS-induced BEAS-2B cells. LPS-induced BEAS-2B cells were transfected with OE-DTX3L or OE-NC. A) Cell viability was assessed using CCK-8 assay. B) Pyroptosis rate was determined using flow cytometry. C) Western blot measured NLRP3, ASC, caspase-1 and GSDMD expression. D) The contents of IL-18 and IL-1β were evaluated by ELISA kits. E) LDH activity was evaluated by a commercial kit. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. DTX3L – Deltex E3 ubiquitin ligase 3 like, OE-DTX3L – overexpression of DTX3L, OE-NC – overexpression negative control, CCK-8 – Cell Counting Kit-8, ASC – apoptosis-associated speck-like protein containing a CARD, GSDMD – gasdermin D, IL-18 – interleukin 18, IL-1β – interleukin 1β, LDH – lactate dehydrogenase
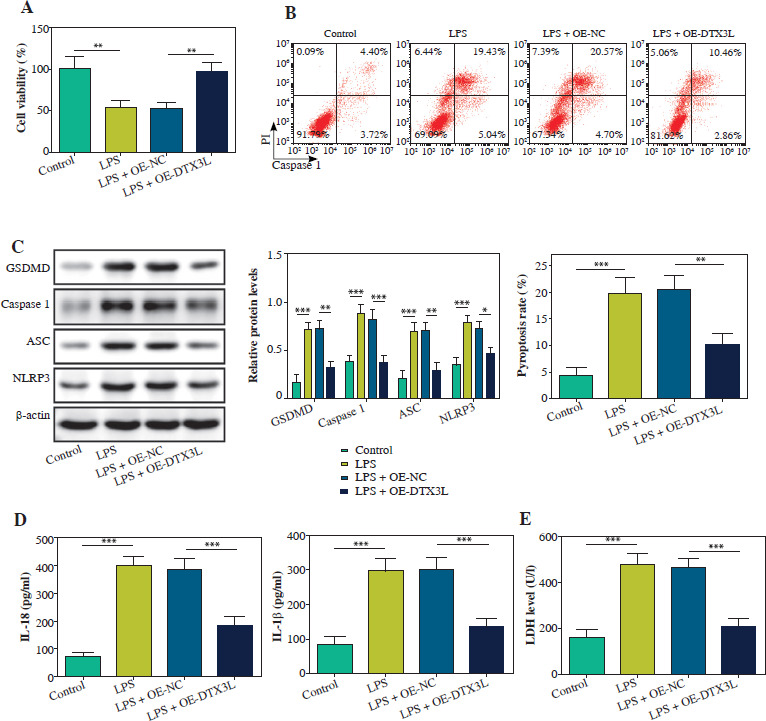
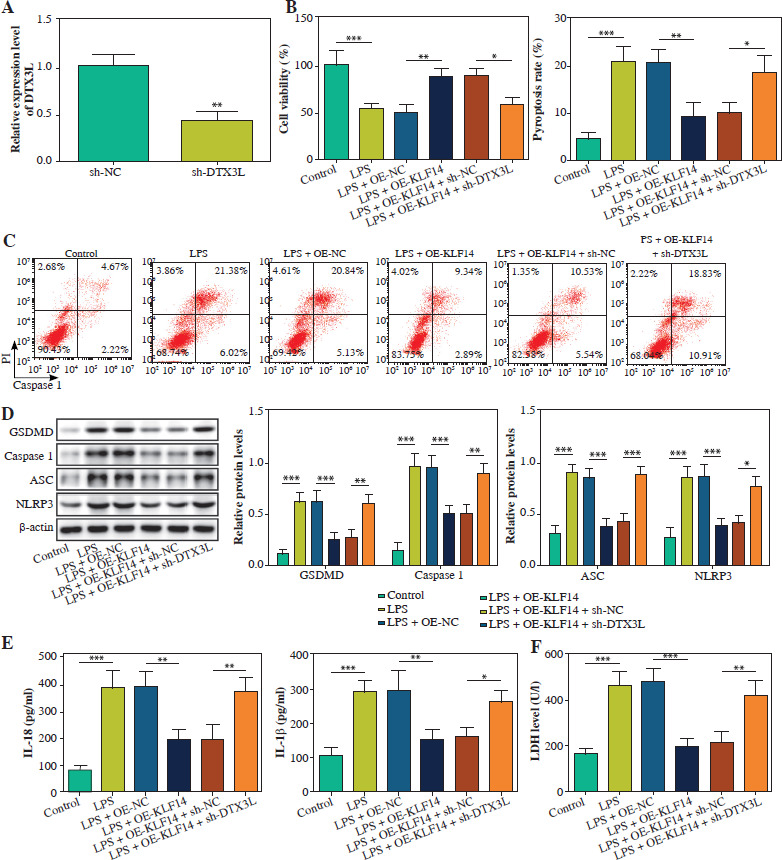
KLF14 suppressed NLRP3-mediated pyroptosis via enhancing DTX3L expression in LPS-induced BEAS-2B cells
Finally, we investigated the KLF14-DTX3L regulatory axis in pyroptosis. In BEAS-2B cells transfected with sh-DTX3L, the DTX3L level was markedly reduced (Fig. 5A). KLF14 upregulation led to a notable increase in cell viability and reduction in pyroptosis, but these effects were abolished by DTX3L depletion in LPS-treated BEAS-2B cells (Fig. 5B, C). Moreover, overexpressing KLF14 repressed the expression of NLRP3, ASC, caspase-1, and GSDMD, and this effect was rescued by depletion of DTX3L in BEAS-2B cells injured by LPS (Fig. 5D). Similarly, KLF14-mediated inhibition of IL-18 and IL-1 β secretion was also negated by DTX3L silencing (Fig. 5E). Furthermore, LDH release was suppressed by KLF14 overexpression but enhanced by co-transfection with sh-DTX3L (Fig. 5F). These findings demonstrated that KLF14 overexpression suppressed LPS-induced pyroptosis by transcriptionally activating DTX3L.
Fig. 5
Upregulation of KLF14 suppressed NLRP3-mediated pyroptosis through enhancing DTX3L expression in LPS-stimulated BEAS-2B cells. A) sh-DTX3L or sh-NC was transfected into BEAS-2B cells. DTX3L mRNA was detected using qRTPCR. LPS-induced BEAS-2B cells were transfected with OE-NC, OE-KLF14, OE-KLF14 + sh-NC, OE-KLF14 + sh-DTX3L. B) CCK-8 assay was used to analyze cell viability. C) Flow cytometry was employed to quantify pyroptosis. D) Protein levels of NLRP3, ASC, caspase-1 and GSDMD were detected using western blot. E) The contents of IL-18 and IL-1β were evaluated by ELISA kits. F) LDH activity was evaluated by a commercial kit. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. DTX3L – Deltex E3 ubiquitin ligase 3 like, sh-DTX3L – short hairpin RNA targeting DTX3L, sh-NC – short hairpin RNA negative control, OE-DTX3L – overexpression of DTX3L, OE-NC – overexpression negative control, qRT-PCR – quantitative reverse transcription polymerase chain reaction, CCK-8 – Cell Counting Kit-8, ASC – apoptosis-associated speck-like protein containing a CARD, GSDMD – gasdermin D, IL-18 – interleukin 18, IL-1β – interleukin 1β, LDH – lactate dehydrogenase
Discussion
Current therapeutic strategies of ALI focus on mitigating inflammation, and reducing oxidative stress and apoptosis in lung tissues [20]. Despite advances in managing sepsis-induced ALI, mortality rates remain unacceptably high [21]. Recently, KLF14 has emerged as a potential regulator of inflammatory pathways [22], but its role in ALI remains underexplored. Previous studies showed that the inhibition of NLRP3-mediated pyroptosis ameliorated sepsis-induced ALI in mice [23]. Nevertheless, the precise mechanisms underlying sepsis-induced ALI still remain uncertain. This study demonstrated that KLF14 inhibited NLRP3-mediated pyroptosis by transcriptionally activating DTX3L, thereby alleviating ALI.
BEAS-2B cells is a human bronchial epithelial cell line widely used in respiratory disease research [24, 25]. LPS-induced BEAS-2B cells can mimic pathological features of sepsis-induced ALI, making them a well-established in vitro model for studying sepsis-induced ALI [26, 27]. Therefore, we used LPS-stimulated BEAS-2B to simulate ALI in vitro models with reference to existing literature. KLF14, a Kruppel-like transcription factor involved in metabolic and immune regulation [28, 29], has shown protective effects in inflammation. For instance, KLF14 mitigated immune-mediated liver injury by suppressing the inflammatory cytokines IL-1 β and IL-18 [30]. The overexpression of KLF14 significantly reduced the production of inflammatory mediators and extracellular matrix (ECM)- degrading enzymes, thereby mitigating osteoarthritis [31]. Another study reported that KLF14 could inhibit the apoptosis of alveolar epithelial cells in an LPS-mediated mouse model of acute lung injury [18]. These studies suggested that KLF14 has suppressive effects on ALI. Aligned with these findings, our results showed that KLF14 upregulation suppressed the secretion of IL-18 and IL-1 β in LPS-stimulated BEAS-2B cells. Pyroptosis, a key contributor to ALI pathogenesis, amplifies inflammation through IL-1 β /IL-18 release, disrupts epithelial barriers, and exacerbates lung injury [32]. The above research indicated a potential crosstalk between KLF14 and pyroptosis. Consistently, our results revealed that KLF14 expression was reduced in LPS-induced cells, and KLF14 overexpression suppressed pyroptosis in an ALI cell model.
KLF14 regulates target genes by binding their promoters. For example, KLF14 transactivated peroxisome proliferator-activated receptor γ (PPAR γ) by binding to its promoter in hepatic stellate cells [33]. Additionally, repressed KLF14 interacted with Wnt family member 3A (WNT3A) promoter in osteogenic differentiation of human bone marrow mesenchymal stem cells [34]. Here, we identified DTX3L as a novel transcriptional target of KLF14, and KLF14 promoted DTX3L transcription. Recent research has highlighted DTX3L’s involvement in inflammation and pyroptosis. Reportedly, DTX3L mitigated RSV-induced bronchiolitis and pneumonia by ubiquitinating TANK-binding kinase 1 (TBK1) [35]. Notably, our results demonstrated that DTX3L inhibited the release of IL-18 and IL-1β in LPS-injured BEAS-2B. The study by Zhou et al. illustrated that DTX3L suppressed NLRP3-mediated pyroptosis to alleviate ischemia/reperfusion injury [14]. As expected, our research showed that KLF14 inhibited cell pyroptosis in ALI by activating DTX3L.
Ubiquitination of NLRP3 may lead to its degradation or alter its ability to interact with other proteins involved in pyroptosis. As reported, tripartite motif-containing protein 31 (TRIM31) increased the ubiquitination level of NLRP3, thereby inhibiting NLRP3 inflammasome and pyroptosis in human retinal pigment epithelium (RPE) cells [36]. Many studies have shown that NLRP3-mediated pyroptosis exacerbates ALI. For instance, blocking p38 MAPK signaling attenuated ALI and excessive lung inflammation by inhibiting NLRP3-mediated pyroptosis [37]. Zhang et al. also found that metformin exhibited a protective role in LPS-induced ALI by decreasing NF-κB-NLRP3-mediated pyroptosis in endothelial cells via increasing sirtuin 1 (SIRT1) expression [38]. Interestingly, DTX3L inhibited OGD/R-induced pyroptosis in R28 cells through regulating NLRP3 ubiquitination [14]. Our study proved that DTX3L promoted NLRP3 ubiquitination and proteasomal degradation, and DTX3L suppressed NLRP3-mediated pyroptosis in LPS-stimulated BEAS-2B cells by promoting NLRP3 ubiquitination, consistent with previous reports.
In summary, our study identified KLF14 as a transcriptional activator of DTX3L. It was demonstrated that overexpression of KLF14 inhibited NLRP3-mediated pyroptosis by activating DTX3L, thereby mitigating sepsis-induced ALI. Our work might offer a promising new clinical target for treating sepsis-induced ALI.