Introduction
Acute lung injury (ALI), characterized by noncardiogenic pulmonary edema and progressive hypoxemia, has garnered significant interest within the realm of critical care medicine due to its high rates of morbidity and mortality [1, 2]. It has been reported that the primary mechanism underlying ALI is an unregulated inflammatory response driven by cytokines such as tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β) and interleukin 6 (IL-6) [3, 4]. An excessive inflammatory response leads to alveolar interstitial destruction and diffuse alveolar epithelial cell damage [5]. Alveolar epithelial cells play a crucial role in safeguarding lung tissue from injury through the secretion of alveolar surfactant, the regulation of ion channels, and the formation of a pulmonary vascular endothelial-alveolar epithelial barrier [6]. If inflammation and epithelial damage are further aggravated, it will develop into acute respiratory distress syndrome (ARDS), bringing life-threatening consequences for patients [7]. Research conducted on animal models of ALI has demonstrated that inhibiting the inflammatory response can effectively mitigate the degree of lung damage [8, 9]. This underscores the importance of investigating the regulatory mechanisms of inflammation in ALI, as it holds considerable implications for the clinical diagnosis and treatment of ALI.
Krüppel-like factor 7 (KLF7) is a DNA-binding transcription factor with a C2H2 zinc finger motif that influences cell proliferation, differentiation, and other cellular functions by regulating the transcription of target genes [10]. However, recent studies have identified KLF7 as an important regulator of inflammation, demonstrating its pro-inflammatory role in various pathological conditions [11, 12]. For example, KLF7 promoted adipocytes to secrete IL-6 by activating the PKCζ/NF-κB pathway, thereby affecting insulin resistance [13]. Huang et al. found that KLF7 was associated with inflammatory accumulation in coronary artery disease, in which KLF7 depletion remarkedly restrained the release of inflammatory factors in lipopolysaccharide (LPS)-induced macrophages [14]. Additionally, the study of Jiang et al. revealed that miR-19b-3p targeted and suppressed KLF7 expression to attenuate the inflammation in sepsis-induced ALI [15]. Nonetheless, the direct role of KLF7 in LPS-induced ALI has not been reported, and its impact on the inflammatory response associated with ALI remains to be explored in our study.
LIM domain kinase 1 (LIMK1) belongs to the LIM kinase protein family [16]. As an important signal transduction protein, it regulates cell morphology and function by phosphorylating and inactivating actin-depolymerizing factors [17]. Esculin was observed to inhibit the PAK1/LIMK1/cofilin pathway to relieve LPS-induced ALI in mice [18]. Similarly, Min et al. reported that inhibition of the Rac/LIMK1 pathway alleviated LPS-induced lung tissue damage and inflammatory cell infiltration [19]. Furthermore, preliminary research has indicated that endotoxin-induced pulmonary edema formation and the infiltration of neutrophils into lung tissues were significantly reduced in LIMK1-deficient mice [20]. The above findings suggest that LIMK1 is involved in the pathogenesis of ALI. Additionally, predictions derived from the JASPAR (https://jaspar.elixir.no/) database revealed that KLF7 has a binding site with the LIMK1 promoter, indicating that KLF7 may enhance the inflammatory response in ALI by activating LIMK1 transcription.
Serine arginine protein kinase 1 (SRPK1) is a protein kinase with a selective pre-mRNA cleavage effect [21]. Apart from its association with cancer progression, numerous studies have confirmed that SRPK1 also contributes to pro-inflammatory processes in multiple diseases [22]. Li et al. found that DLEU1 upregulated the expression of SRPK1, thereby promoting inflammation of the spinal cord in chronic constrictive injury animal models [23]. Moreover, SRPK1 was reported to promote sepsis-induced ALI by regulating the PI3K/AKT/FOXO3 pathway [24]. Additionally, the research of Yao et al. showed that SRPK1 was highly expressed in LPS-induced ALI and aggravated the inflammatory response by regulating AKT3 [25]. Interestingly, we used Hitpredict (https://www.hitpredict.org/) and BioGRID (https://thebiogrid.org/) databases and predicted that LIMK1 might have a protein interaction possibility with SRPK1. A study on breast cancer showed that LIMK2 effectively impeded the phosphorylation and functional activity of SRPK1 [26]. Therefore, we aimed to determine whether LIMK1 can enhance the phosphorylation and activity of SRPK1 and subsequently influence the inflammatory response under ALI conditions.
Building upon the aforementioned information, we hypothesize that KLF7 may transcriptionally modulate LIMK1, which in turn promotes the phosphorylation and activation of SRPK1, thereby amplifying the inflammatory response of alveolar epithelial cells in the context of ALI. This investigation will hopefully offer novel insights into the pathogenesis of ALI and inform the development of targeted therapy strategies.
Material and methods
Cell culture and treatment
The human primary type II alveolar epithelial cell line (AT II) was obtained from Procell Co. Ltd (CP-H209, China). F12 medium (Gibco, USA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution was applied to maintain cell growth. Cells were placed in a humidified incubator at 37oC with 5% CO2, and the culture medium was refreshed every 2 to 3 days.
To establish ALI in vitro, AT II cells were treated with LPS (10 µg/ml, HY-D1056, MedChemExpress, USA) for 24 h and then replaced with a normal medium [27]. SRPIN340 (an SRPK1 inhibitor) was used to determine the role of SRPK1 in ALI, by which AT II cells were pretreated with SRPIN340 (5, 10, 15, and 20 µM, HY-13949, MedChemExpress, USA) for 24 h before subsequent experiments were performed.
Cell transfection
The short hairpin RNAs (shRNAs) against KLF7 and LIMK1 expression were constructed by GenePharma Co. Ltd (China), as well as the overexpression plasmid of KLF7 and LIMK1. Specifically, the AT II cells were pre-seeded in a 6-well plate, and the transfection process started once the cell confluence was around 60-70%. All the plasmids packaged with lentivirus were transfected into AT II cells using Lipofectamine 3000 reagent (L3000008, Invitrogen, USA) according to the kit instructions [28]. In brief, Lipofectamine 3000 was diluted with Opti-MEM medium. Subsequently, the plasmid DNA was mixed with an appropriate quantity of Opti-MEM medium and P3000 reagent. Following the combination of the two reagent solutions, the resulting mixture was administered to the cells. After 24 h of transfection, 3 µg/ml of puromycin (P8833, Sigma Aldrich, USA) was used to select cells, and then the transfection efficiency was tested for subsequent experiments.
Real-time quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted from cell samples using an RNA isolator (R401-01, Vazyme, China). The HiScript II 1st Strand cDNA Synthesis Kit (R212-01, Vazyme, China) was employed to reverse transcribe the extracted RNA into complementary DNA (cDNA). Following this, RNase-free ddH2O was used to prepare a 20 µl reaction mixture for real-time PCR (Roche LightCycler 96, Switzerland), which included the cDNA template, primers, and 2×SuperReal PreMix Plus (FP205-01, TIANGEN, China). β-actin was designated as the internal reference gene to compare gene expression variations. The primer sequences used in this study were as follows (5´-3´):
KLF7 Forward: AGACATGCCTTGAATTGGAACG
KLF7 Reverse: GGGGTCTAAGCGACGGAAG
LIMK1 Forward: CAAGGGACTGGTTATGGTGGC
LIMK1 Reverse: CCCCGTCACCGATAAAGGTC
SRPK1 Forward: ATGGAGCGGAAAGTGCTTG
SRPK1 Reverse: GAGCCTCGGTGCTGAGTTT
TNF-α Forward: CCCCAGGGACCTCTCTCTAA
TNF-α Reverse: TGAGGTACAGGCCCTCTGAT
IL-1β Forward: ATGATGGCTTATTACAGTGGCAA
IL-1β Reverse: GTCGGAGATTCGTAGCTGGA
IL-6 Forward: ACAGGGAGAGGGAGCGATAA
IL-6 Reverse: GAGAAGGCAACTGGACCGAA
β-actin Forward: TGGCACCACACCTTCTACAA
β-actin Reverse: CCAGAGGCGTACAGGGATAG
Western blot
To obtain the total protein of cell samples, RIPA lysis buffer (P0013B, Beyotime, China) mixed with protease and phosphatase inhibitors was used. After quantifying the protein concentration using a BCA analysis kit (P0010S, Beyotime, China), an appropriate volume of sodium dodecyl sulfate (SDS) loading buffer (P0015L, Beyotime, China) was added to the protein sample. Subsequently, an SDS-PAGE gel was prepared for electrophoretic separation. After the separation process, the proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (88518, Thermo Scientific, USA). The membrane was then soaked and blocked in 5% skim milk for 1 h at room temperature. Following this, the membrane was respectively exposed to the primary antibodies against KLF7 (sc-398576, Santa Cruz Biotechnology, USA), LIMK1 (ab95186, Abcam, UK), SRPK1 (ab90527, Abcam, UK), p-SRPK1 (PA5-106156, Invitrogen, USA), and β-actin (ab8227, Abcam, UK) for overnight incubation at 4oC. After the removal of the primary antibody, the membrane was treated with the horse radish peroxidase (HRP)-conjugated secondary antibodies (ab288151, ab6728, Abcam, UK). Finally, the proteins were visualized with the help of ECL chromogenic solution under an imaging system (Azure 600, USA), and the results were analyzed using ImageJ (version: 1.8.0, National Institutes of Health, USA).
Enzyme-linked immunosorbent assay (ELISA)
In brief, the collected cell supernatant and biotin- labeled antibody were incubated simultaneously. After washing, avidin-labeled HRP was added. Following another incubation and washing phase, substrates A and B were introduced with the concurrent action of the enzyme conjugate. Finally, the sample to be tested was placed into a microplate reader (Thermo Fisher Scientific, USA) to detect absorbance at 450 nm. The ELISA kits of TNF-α (ml077385), IL-1β (ml058059), and IL-6 (ml058097) were purchased from Enzyme-linked Biotechnology Co., Ltd (China).
CCK-8 assay
The Cell Counting Kit-8 (CCK-8) assay was employed to assess alterations in the viability of AT II cells after treatment with LPS and/or transfection with related plasmids. Briefly, AT II cells were seeded into a 96-well plate, with an approximate density of 2 × 104 cells per well. 24 hours later, 10 µl of CCK-8 reagent (C0037, Beyotime, China) was added to each well for assessment. The wells were then incubated at 37oC for an additional hour, after which the absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, USA).
Flow cytometry for cell apoptosis detection
Cells were harvested using ethylenediaminetetraacetic acid (EDTA)-free trypsin digestion. Following a washing step, the cells were resuspended in a binding solution, to which Annexin V-FITC (C1062S, Beyotime, China) and propidium iodide (C1062S, Beyotime, China) were added and thoroughly mixed. The mixture was incubated at room temperature without light for at least 20 min. Subsequently, flow cytometry (Beckman Coulter, USA) was used to assess cell apoptosis within one hour, and the data were analyzed using FlowJo software (version: 10.8.1, Tree Star, USA).
Dual-luciferase reporter assay
The wild-type and mutant LIMK1 promoter fragments were inserted into the pGL3-basic vector (Promega, USA) to generate various dual-luciferase reporter plasmids. Subsequently, the constructed plasmid vector was co-transfected into AT II cells alongside either the sh-NC or sh-KLF7 plasmid. Twenty-four hours after transfection, firefly and Renilla luciferase activities were measured using the Dual- Glo system (E2920, Promega, USA), and the ratio of luciferase activities between the two groups was calculated to assess transcriptional activity.
Chromatin immunoprecipitation (ChIP)
This assay applied the Pierce Magnetic ChIP Kit (26157, Thermo Fisher Scientific, USA) [29]. In summary, cells were treated with a culture medium containing a 1% formaldehyde solution at 37oC for 10 minutes, after which the cross-linking reaction was halted using glycine. Subsequently, the cells were lysed and the chromatin was fragmented using a sonicator. Following this, the nuclear pellet was resuspended in an IP dilution buffer, and either IgG (ab172730, Abcam, UK) or KLF7 antibody (sc-398576, Santa Cruz Biotechnology, USA) was introduced and incubated overnight at 4oC. After incubation, 20 µl of magnetic beads were added to each IP sample and incubated for an additional 2 hours at 4oC with gentle mixing. The washed beads were then eluted using IP elution buffer to isolate the DNA-protein complexes. Finally, purified DNA was obtained through a DNA Recovery process, allowing for subsequent qPCR analysis.
Glutathione-S-transferase (GST) pull-down assay
The plasmid with the GST tag and SRPK1 sequence was introduced into BL21 competent cells, which were cultured overnight. The bacterial culture from the positive clone was then expanded. Following the collection of the bacterial cells, they were resuspended in a buffer supplemented with protease inhibitors, and bacterial lysis was achieved through sonication. The supernatant was collected after centrifugation. The prepared cell lysate was then combined with the GST fusion bait protein and incubated at 4oC with gentle agitation overnight. The following day, glutathione-agarose beads were added to the mixture, which was shaken and incubated for 4 hours at 4oC. The beads were washed repeatedly, and the interacting protein complex was subsequently eluted from the beads using an elution buffer. Verification and identification of the eluted proteins were conducted using Western blot analysis.
Co-immunoprecipitation (Co-IP)
This experiment was carried out using the Pierce Classic Magnetic IP/Co-IP Kit (88804, Thermo Fisher Scientific, USA) [30]. In brief, AT II cells were treated with RIPA lysis buffer at a low temperature for 30 min to isolate the protein. Subsequently, the cell lysates were incubated overnight at 4oC with IgG (ab172730, Abcam, UK) or the antibody against LIMK1 (ab95186, Abcam, UK) to ensure complete binding. The following day, the protein-antibody mixture was added to a centrifuge tube containing Protein A/G beads and incubated at room temperature for 2 hours. The beads were then collected using a magnetic stand, and unbound samples were removed. The magnetic beads were washed multiple times using IP lysis/wash buffer, and the precipitate was eluted using an elution buffer. The obtained samples were analyzed through immunoblotting experiments to assess protein-protein interactions.
Statistical analysis
All data are presented as mean ± standard deviation, and the analytical procedures were conducted using GraphPad Prism 9.0 software (version: 9.0, GraphPad Software, USA). Student’s t-test was employed to assess differences in continuous variables between two groups. For comparisons involving more than two groups, one-way analysis of variance (ANOVA) followed by Tukey’s test was used. Each experiment was conducted three times to ensure reliability. A p-value of less than 0.05 was deemed statistically significant.
Results
Knockdown of KLF7 inhibited LPS-induced inflammatory response and apoptosis of AT II cells
Our initial study found that the mRNA and protein levels of KLF7 were elevated in AT II cells treated with LPS (Fig. 1A, B). To confirm the role of KLF7, we further constructed a KLF7 knockdown vector and infected it into AT II cells. The results, shown in Figure 1C, D, illustrated the efficiency of KLF7 knockdown. On this basis, we further induced ALI in cells. As displayed in Figure 1E, F, KLF7 inhibition significantly counteracted the increased KLF7 expression induced by LPS treatment in AT II cells. LPS treatment caused an increase in the expression and release levels of the inflammatory mediators TNF-α, IL-1β, and IL-6, and their levels decreased when KLF7 declined (Fig. 1G, H). Furthermore, the viability of AT II cells was suppressed by LPS treatment, and the reduction of KLF7 attenuated the observed decrease in cell viability (Fig. 1I). Finally, as shown in Figure 1J, KLF7 suppression also reversed the effect of elevated cell apoptosis induced by LPS treatment. These results demonstrated that KLF7 has a pro-inflammatory function in LPS-induced AT II cells.
Fig. 1
Knockdown of KLF7 inhibited LPS-induced inflammatory response and apoptosis of AT II cells. A, B) AT II cells were treated with LPS (10 μg/ml) for 24 h to induce ALI conditions. RT-qPCR and Western blot were applied to detect KLF7 expression. C, D) sh-KLF7 and its control sh-NC were respectively transfected into AT II cells. RT-qPCR and Western blot analysis were conducted to check the efficiency of KLF7 knockdown. After the transfection of sh-KLF7 and sh-NC, AT II cells were treated with LPS. E, F) RT-qPCR and Western blot were used to measure KLF7 levels. G) RT-qPCR was used to measure the expression levels of TNF-α, IL-1β, and IL-6. H) The secretion levels of TNF-α, IL-1β, and IL-6 were tested using their own ELISA kits. I) CCK-8 assay was applied to determine the viability of AT II cells. J) Flow cytometry was used to quantify cell apoptosis levels. All experiments were conducted in triplicate. **p < 0.01, ***p < 0.001
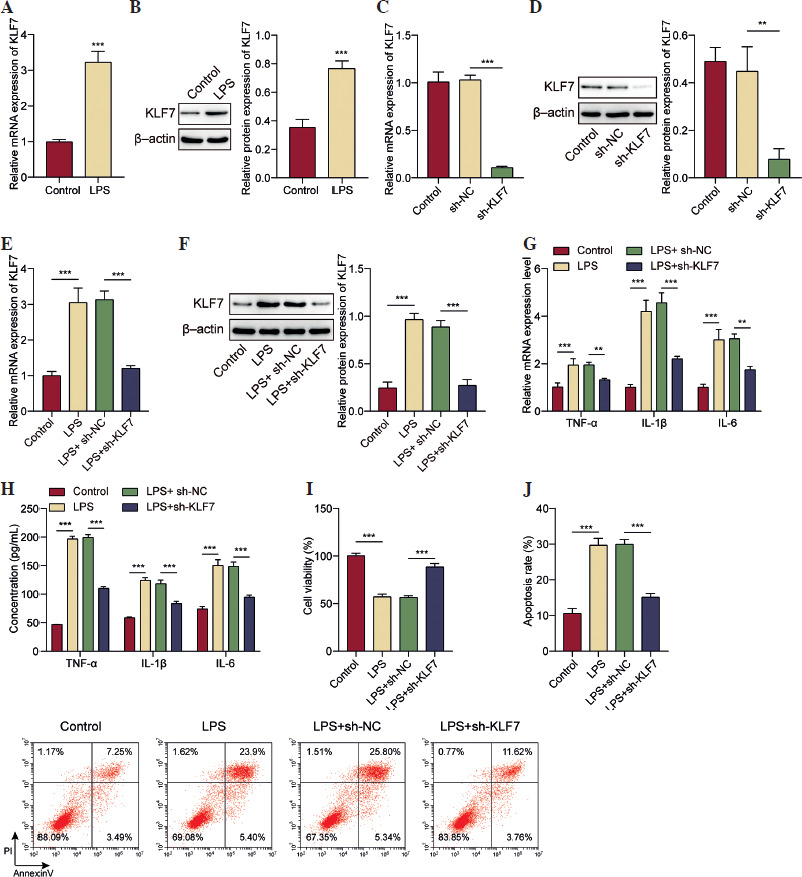
KLF7 promoted LIMK1 transcriptional activation in AT II cells
As illustrated in Figure 2A, B, both mRNA and protein expressions of LIMK1 were increased under ALI conditions. Subsequently, we investigated the potential regulatory interaction between KLF7 and LIMK1. The results, shown in Figure 2C, display the binding sites between the KLF7 and LIMK1 promoter, which were predicted using the JASPAR database. Dual-luciferase assay showed that KLF7 knockdown resulted in a marked reduction in the transcriptional activity of LIMK1, which was abolished when LIMK1 was mutated (Fig. 2D). In addition, KLF7 was greatly enriched in the promoter region of LIMK1 (Fig. 2E). The above results indicated that KLF7 and LIMK1 promoters have a mutual binding relationship. Subsequently, we investigated the effect of KLF7 interference on LIMK1 levels. As illustrated in Figure 2F, G, oe-KLF7 significantly increased KLF7 mRNA and protein levels. KLF7 overexpression increased the mRNA and protein levels of LIMK1 (Fig. 2H, I). In contrast, knocking down KLF7 significantly reduced LIMK1 levels (Fig. 2J, K). As depicted in Figure 2L, M, LIMK1 was found to be upregulated following treatment with LPS, whereas the knockdown of KLF7 abolished this increase. The above findings suggested that KLF7 can bind to the LIMK1 promoter region and then enhance LIMK1 transcription and translation in AT II cells.
Fig. 2
KLF7 promoted LIMK1 transcriptional activation in AT II cells. A, B) AT II cells were treated with LPS (10 μg/ml) for 24 h to induce ALI conditions. RT-qPCR and Western blot were applied to evaluate LIMK1 expression. C) JASPAR database predicted the potential binding site of KLF7 and the LIMK1 promoter. D) Dual-luciferase reporter assay was conducted to test the transcriptional activity of LIMK1 after KLF7 knockdown. E) ChIP experiment was used to confirm the interaction between KLF7 and the LIMK1 promoter. oe-KLF7 and its corresponding control plasmids were respectively transfected into AT II cells. F, G) KLF7 overexpression effect was analyzed by RT-qPCR and Western blot. H, I) LIMK1 expression was assessed by RT-qPCR and Western blot. J, K) LIMK1 expression was assessed by RT-qPCR and Western blot after AT II cells were transfected with sh-KLF7 and its corresponding control plasmids. L, M) AT II cells were treated with LPS after transfection of sh-KLF7. LIMK1 expression was quantified using RT-qPCR and Western blot. All experiments were conducted in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001
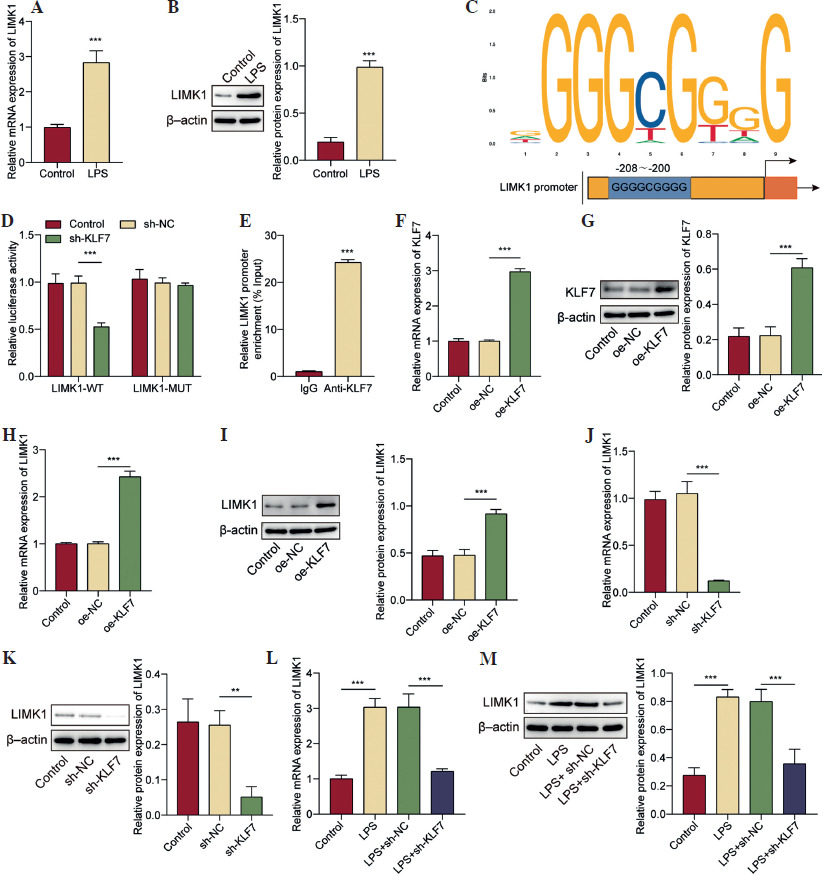
LIMK1 overexpression reversed the effect of KLF7 silencing on LPS-induced inflammatory response and apoptosis of AT II cells
Subsequently, by overexpressing LIMK1 in AT II cells, we explored the role of the KLF7/LIMK1 pathway in ALI. As shown in Figure 3A, B, the transfection of LIMK1 overexpression plasmids resulted in a substantial increase in LIMK1 expression within AT II cells. The increase in LIMK1 levels induced by LPS was eliminated by KLF7 downregulation, and overexpression of LIMK1 further reversed the impact of KLF7 knockdown on LIMK1 expression (Fig. 3C, D). Similarly, LIMK1 overexpression also affected the inflammatory response in AT II cells after LPS treatment. The results, shown in Figure 3E, F, revealed that LIMK1 upregulation elevated the reduced TNF-α, IL-1β, and IL-6 levels caused by KLF7 downregulation. Moreover, the enhanced viability of AT II cells associated with KLF7 deficiency was subsequently diminished by LIMK1 elevation (Fig. 3G). Concurrently, LIMK1 upregulation was found to abolish the influence of KLF7 knockdown on the apoptosis of AT II cells, that is, to exacerbate the apoptosis in these cells (Fig. 3H). This evidence indicated that KLF7 upregulates LIMK1 to facilitate the inflammatory response and apoptosis of AT II cells induced by LPS.
Fig. 3
LIMK1 overexpression reversed the effect of KLF7 silencing on LPS-induced inflammatory response and apoptosis of AT II cells. A, B) LIMK1 was overexpressed in AT II cells. LIMK1 overexpression efficiency was verified by RT-qPCR and Western Blot. KLF7 was knocked down or/and LIMK1 was overexpressed in ALI cell models. C, D) RT-qPCR and Western Blot were applied to assess LIMK1 expression. E) RT-qPCR was utilized to determine TNF-α, IL-1β and IL-6 expressions. F) ELISA assays were applied to measure TNF-α, IL-1β and IL-6 secretion. G) CCK-8 assay was conducted to evaluate cell viability. H) Detection of cell apoptosis was performed by flow cytometry. All experiments were conducted in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001
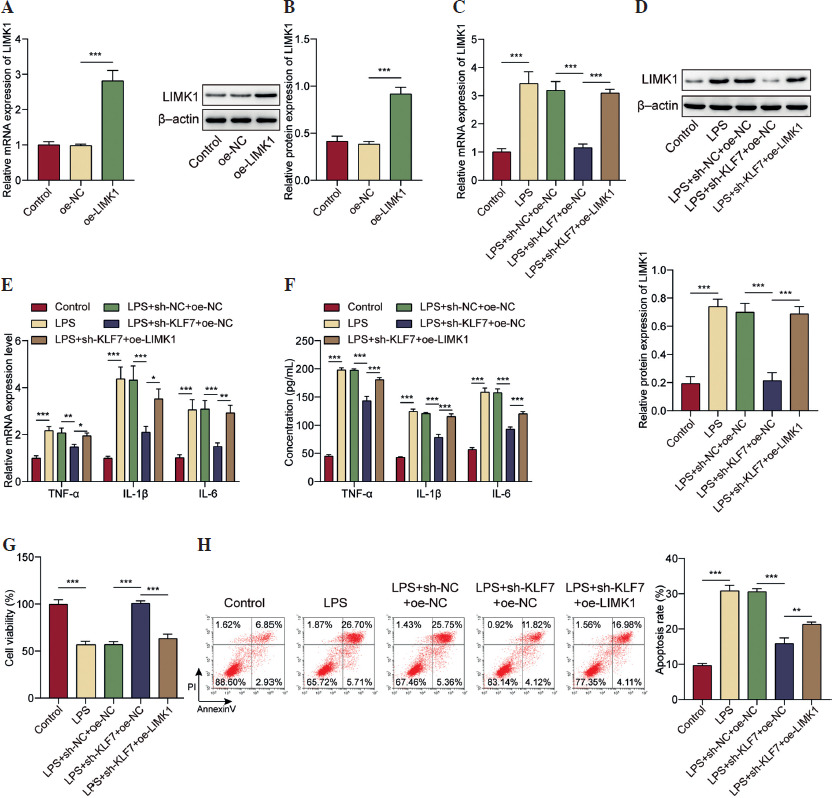
LIMK1 promoted phosphorylation of SRPK1
We continued to investigate the downstream molecules associated with LIMK1 in the context of ALI. Using the HitPredict and BioGRID databases, we predicted a potential interaction between the LIMK1 and SRPK1 proteins. SRPK1 has been proven to act as a pro-inflammatory factor in ALI [25]. LIMK1 is known to regulate protein phosphorylation levels as a phosphokinase [31]. Thus, we further detected changes in SRPK1 protein phosphorylation levels under LPS treatment. As shown in Figure 4A, an increase in both the protein levels and phosphorylation of SRPK1 in LPS-treated AT II cells was observed. Next, we validated the interaction between LIMK1 and SRPK1 through GST pull-down and Co-IP assays (Fig. 4B, C). Following this, we performed a knockdown of LIMK1 in AT II cells. As illustrated in Figure 4D, E, LIMK1 silencing caused a significant reduction in LIMK1 expression. Western blot analysis revealed that the knockdown of LIMK1 specifically affected the phosphorylation level of SRPK1 without altering its overall protein expression (Fig. 4E). Further experiments involving LPS treatment demonstrated that LPS enhanced the phosphorylation activity of SRPK1, and LIMK1 inhibition mitigated the increase in SRPK1 phosphorylation induced by LPS (Fig. 4F). Collectively, LIMK1 interacts with SRPK1 protein in AT II cells to promote its phosphorylation.
Fig. 4
LIMK1 promoted the phosphorylation of SRPK1. A) AT II cells were treated with LPS. Western blot determined the protein and phosphorylation levels of SRPK1. B, C) GST pull-down and Co-IP assays were applied to validate the protein binding relationship between LIMK1 and SRPK1. LIMK1 was silenced in AT II cells. D) RT-qPCR was employed to assess LIMK1 knockdown efficiency. E) Western blot analysis for LIMK1, SRPK1 expression and SRPK1 phosphorylation level. F) LIMK1 was silenced in LPS-treated AT II cells. Western blot was performed to quantify LIMK1, SRPK1 and SRPK1 phosphorylation levels. All experiments were conducted in triplicate. **p < 0.01, ***p < 0.001
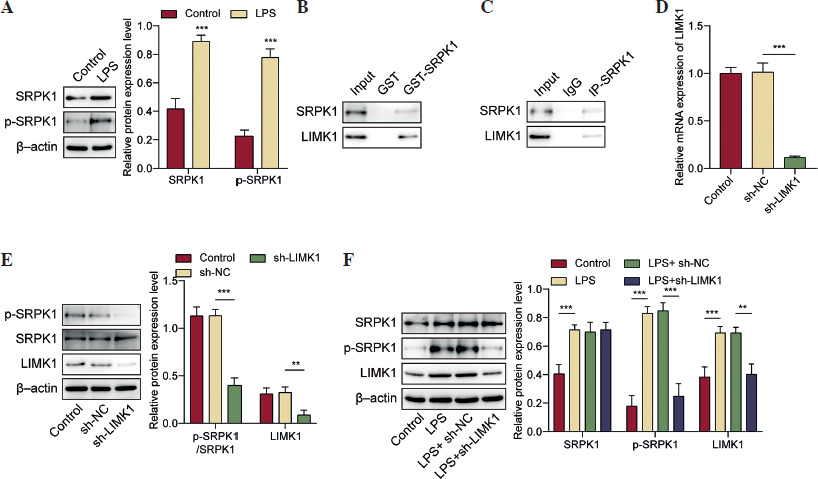
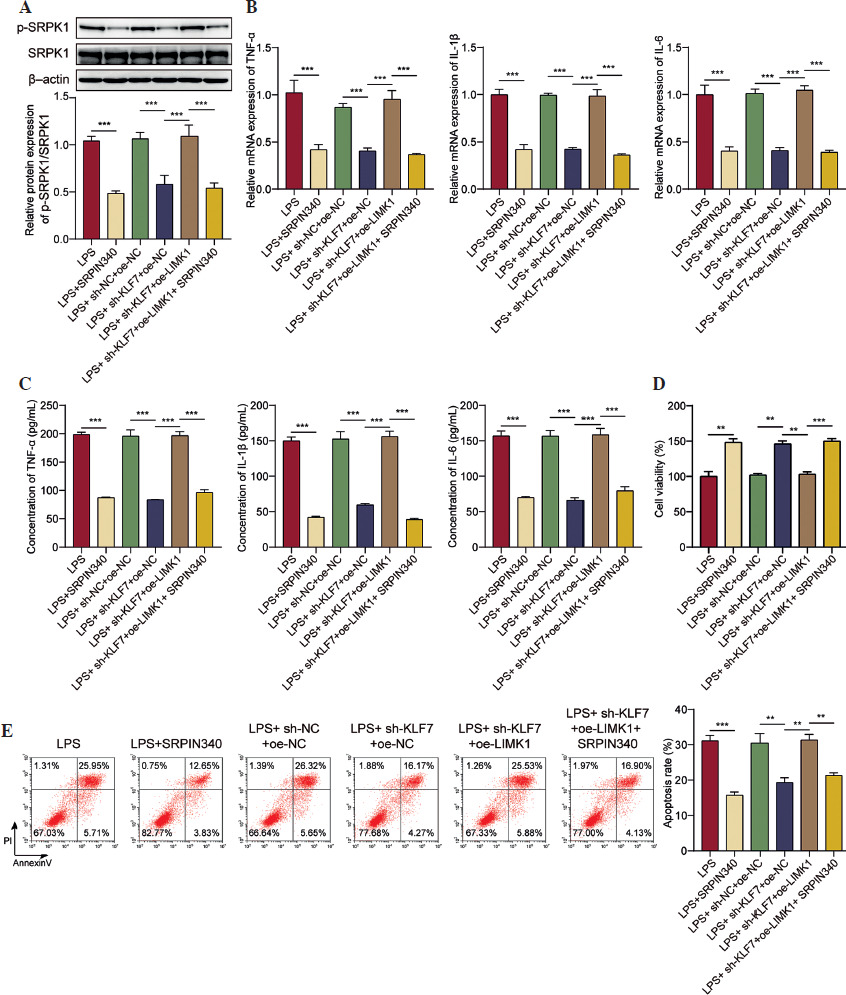
KLF7 stimulated the LPS-induced inflammatory response and cell apoptosis by regulating the LIMK1/SRPK1 axis
Finally, we identified the regulatory function of the KLF7/LIMK1/SRPK1 pathway in ALI. First, the influence of SRPIN340 (SRPK1 inhibitor) on cellular viability was assessed. We observed that 5 µM and 10 µM of SRPIN340 did not produce a significant effect on cell viability. When the concentration reached 15 µM, it reduced cell viability (Suppl. Fig. 1). Since 10 µM SRPIN340 has no cytotoxic effect, this concentration was used for further exploration. As shown in Figure 5A, SRPIN340 reduced SRPK1 phosphorylation in LPS treated AT II cells; LIMK1 upregulation counteracted the inhibitory impact of KLF7 knockdown on SRPK1 activity, while SRPIN340 abolished the effects of LIMK1 overexpression. Furthermore, KLF7 knockdown and SRPIN340 treatment resulted in a reduction in the expression and secretion of inflammatory mediators, respectively. Conversely, the overexpression of LIMK1 antagonized the impact of KLF7 knockdown, while SRPIN340 further mitigated the effects induced by LIMK1 overexpression (Fig. 5B, C). In addition, overexpression of LIMK1 reversed the enhancement effect of KLF7 knockdown on cell viability, while inhibition of SRPK1 further antagonized the decreased effect of overexpression of LIMK1 on cell viability (Fig. 5D). Then flow cytometry was employed to assess cell apoptosis. As illustrated in Figure 5E, SRPIN340 suppressed the cell apoptosis induced by LPS; moreover, LIMK1 upregulation was observed to counteract the anti-apoptotic effects resulting from KLF7 knockdown, and SRPIN340 abolished the impact of LIMK1 overexpression again. In conclusion, KLF7 aggravated the inflammatory response and apoptosis in LPS-treated AT II cells via the LIMK1/SRPK1 pathway.
Fig. 5
KLF7 stimulated LPS-induced inflammatory response and cell apoptosis by regulating the LIMK1/SRPK1 axis. AT II cells were treated with sh-KLF7, oe-LIMK1, and/or SRPIN340, followed by LPS treatment. A) Western blot was applied to determine the phosphorylation of SRPK1. B) RT-qPCR was used to detect the expression TNF-α, IL-1β, and IL-6. C) ELISA assays were applied to assess the secretion of TNF-α, IL-1β, and IL-6. D) Cell viability was evaluated by CCK-8 assay. E) Cell apoptosis was measured by flow cytometry. All experiments were conducted in triplicate. **p < 0.01, ***p < 0.001
Discussion
In the case of ALI, the inflammatory response triggers the release of numerous inflammatory mediators, resulting in the excessive recruitment of immune cells [32]. This process ultimately leads to the destruction of the alveolar epithelial cell barrier and subsequent cell death [33]. The ongoing damage to the epithelium creates opportunities for the invasion of additional bacteria and viruses, which is a critical factor in the progression of ALI to ARDS and the exacerbation of the condition [7]. Our research has identified a novel mechanism that enhances the inflammatory response and contributes to alveolar epithelial cell damage in ALI. Specifically, KLF7 activates the transcription of LIMK1, and the upregulation of LIMK1 facilitates the phosphorylation of SRPK1, thereby intensifying the inflammatory response and alveolar epithelial cell damage in LPS-induced alveolar epithelial cells.
The transcription factor KLF7 is widely expressed in various tissues and can bind to the promoter regions of CACCC/GC-rich genes, thereby modulating the expression of downstream genes [34]. For instance, KLF7 facilitated the secretion of pro-inflammatory cytokines in rheumatoid arthritis (RA) by activating the NF-κB and JNK pathways [12]. KLF7 has also been shown to promote the transcription of the essential inflammatory mediator IL-6 directly [35]. Our research found that KLF7 was elevated in LPS-induced ALI, indicating its potential regulatory role. LPS, a main constituent of the outer membrane of Gram-negative bacteria, is recognized as a primary contributor to ALI [33]. LPS engages and activates toll-like receptor 4, initiating an inflammatory response that sensitizes immune cells and increases the permeability of the alveolar-capillary membrane, ultimately leading to lung tissue damage [36]. In our investigation, the knockdown of KLF7 markedly reduced the expression and secretion of the inflammatory mediators TNF-α, IL-1β, and IL-6 and apoptosis in alveolar epithelial cells induced by LPS. These results prove, for the first time that, KLF7 functions as a pro-inflammatory factor in LPS-induced ALI.
LIMK1 is crucial for alternating cytoskeletal actin dynamics and the depolymerization of microtubules, thereby regulating multiple cell functions [16]. It was found that serine protease A facilitated the invasion of Shigella into the intestinal epithelium and elicited a significant inflammatory response through the modulation of cofilin protein expression, which is mediated by LIMK1 [37]. The downregulation of LIMK1 has been shown to mitigate the inflammatory response in murine models by decreasing actin polymerization and the formation of focal adhesions [38]. This evidence underscores the critical role of LIMK1 in the pathophysiology of inflammation-related disorders. Notably, LIMK1 has been demonstrated to exert a pro- inflammatory effect in animal models of LPS-induced ALI [18, 19]. Our research revealed that LIMK1 was upregulated in response to LPS stimulation, and we successfully identified a new upstream regulator of LIMK1 in alveolar epithelial cells: KLF7, which binds to the LIMK1 promoter, enhancing its transcription. Furthermore, subsequent rescue experiments indicated that LIMK1 overexpression mitigated the inhibitory effects of KLF7 knockdown on the LPS-induced inflammatory response and cell apoptosis. This suggests that KLF7 and LIMK1 constitute a signaling pathway that regulates the pathogenesis of ALI.
As a serine/threonine kinase, the major function of LIMK1 is to phosphorylate the protein cofilin, therefore participating in the development of several cancers and diseases [31, 39]. Research conducted by Chen et al. demonstrated that LIMK1 mediated the phosphorylation of eIF2α in colorectal cancer [40]. Similarly, LIMK1 was found to enhance the phosphorylation of SRPK1 in our study. SRPK1 is a serine-arginine protein kinase, and the alterations in its phosphorylation status can affect its function as a splicing regulator. This, in turn, may influence the expression or phosphorylation of several factors, including AKT, NF-κB, and TGF-β [39]. A study revealed that SRPK1 was involved in sevoflurane-induced neuroinflammatory processes and neuronal apoptosis [22]. More importantly, Guo et al. and Yao et al. reported that SRPK1 promoted sepsis or LPS-induced ALI [24, 25]. Here, we presented the novel finding that KLF7 activated SRPK1 through upregulating LIMK1, a process that aggravated the inflammatory response and apoptosis in alveolar epithelial cells treated with LPS. However, we also observed that LIMK1 specifically influenced the phosphorylation of SRPK1, while LPS appeared to elevate the overall protein levels of SRPK1. The mechanisms underlying the upregulation of SRPK1 protein levels in ALI warrant further exploration.
Nevertheless, our research is not without limitations. Specifically, our hypothesis has only undergone preliminary validation at the cellular level, necessitating additional animal and clinical studies for comprehensive confirmation. In the future, we aim to examine the upstream mechanisms of KLF7 and identify the specific phosphorylation sites of SRPK1 to improve our mechanistic hypothesis. Overall, our study demonstrates that KLF7 transcriptionally activates LIMK1, which subsequently enhances the phosphorylation of SRPK1, thereby amplifying the inflammatory response of alveolar epithelial cells under ALI conditions. This evidence will provide a foundational laboratory basis for understanding the pathogenesis of ALI and for developing targeted therapeutics.