Introduction
Acute lung injury (ALI) is a prevalent clinical condition that poses a significant threat to respiratory function and life. It is characterized by inflammation, cellular damage, and elevated alveolar permeability of lung tissues, resulting in diminished lung functionality and oxygenation [1]. Progression of ALI might subsequently result in acute respiratory distress syndrome (ARDS) [1]. Despite notable advancements in the clinical recognition and management of ARDS, the mortality rate remains approximately 30% to 40% [2]. The endoplasmic reticulum (ER) is responsible for synthesizing, folding, and repairing proteins. However, when this function is compromised, the accumulation of misfolded or unfolded proteins occurs, which triggers endoplasmic reticulum stress (ERS) [3]. It was demonstrated that ERS was critically involved in the pathogenesis of ALI. The level of ERS and the expression of C/EBP homologous protein (CHOP) were discovered to be markedly elevated in the ALI model induced by lipopolysaccharide (LPS) [4]. The impairment of ERS-induced apoptosis in alveolar epithelial cells (AECs) effectively ameliorated ALI caused by myocardial ischemia-reperfusion [5]. It could be reasonably proposed that modulating ERS in AECs might be a novel therapeutic approach for the management of ALI.
Casitas B lymphoma-b (CBLB), a pivotal regulator of human autoimmune diseases, was documented to exert tumor-suppressive effects by enhancing anti-tumor T-cell responses [6]. CBLB protein contains four structural domains, including ubiquitin-associated structural domains, and has been widely documented to facilitate the degradation of downstream target proteins in a proteasome-dependent manner [7]. A study demonstrated that the increased expression of CBLB enhanced the protein degradation of mitogen-activated protein kinase kinase 4 in pancreatic β-cells, which diminished the level of cellular ERS-induced c-Jun N-terminal kinase (JNK) phosphorylation [8]. Notably, the absence of CBLB expression caused an increase in sepsis-induced release of inflammatory cytokines and chemokines, which exacerbated lung inflammation in ALI [9]. Nevertheless, whether CBLB participates in the modulation of ERS in ALI remains uninvestigated.
N6-methyladenosine (m6A) represents the most prevalent type of RNA methylation modification observed in eukaryotic organisms, occurring at the N6 position of adenosine. It plays a pivotal role in regulating gene expression [10]. The reversible process of m6A modification is carried out by both methyltransferases and demethylases. RNAs with m6A modifications are identified by m6A reader proteins, which ultimately determine their fate [11]. It was demonstrated that significantly elevated overall m6A modifications in AECs were closely correlated with disease progression of ALI. For example, methyltransferase-like 14 (METTL14), a crucial methyltransferase, was reported to exacerbate autophagy in AECs subjected to ALI [12]. Moreover, it was reported that knockdown of methyltransferase-like 3 (METTL3) enhanced glutathione peroxidase 4 (GPX4) expression to attenuate ferroptosis in sepsis-induced ALI [13]. Intriguingly, the prediction by the SRAMP (http://www.cuilab.cn/sramp) database showed multiple m6A modification sites on CBLB mRNA. Furthermore, the mRNA of CBLB showed the potential to bind to multiple m6A readers, such as YTH N6-methyladenosine RNA binding protein F2 (YTHDF2) and YTH N6-methyladenosine RNA binding protein C1 (YTHDC2). Therefore, we speculated that the abnormal expression of CBLB in ALI might be regulated by m6A modification.
LIM domain kinase 1 (LIMK1) is a serine/threonine kinase that exerts biological function by phosphorylating downstream target proteins [14]. LIMK1 was reported to participate in various inflammation-related disease processes, and enhancement of LIMK1-mediated transduction of the cofilin/actin signaling pathway facilitated the regulation of the endothelial cytoskeleton and augmented inflammation-induced cellular permeability [15]. Importantly, LIMK1 exacerbated lung endothelial barrier injury and enhanced neutrophil infiltration in mice with ALI, and LIMK1 inhibition was observed to alleviate ALI [16]. Additionally, it was found that LIMK1 overexpression in colorectal cancer cells promoted eukaryotic translation initiation factor 2α (eIF2 α) phosphorylation to facilitate ERS [17]. Furthermore, we predicted that LIMK1 might be a substrate for CBLB to exert ubiquitination through the UbiBrowser (http://ubibrowser.bio-it.cn/ubibrowser/home/index) database. However, it remains unclear whether CBLB exerts a regulatory effect on the occurrence of ERS in AECs during ALI by modulating the ubiquitination degradation of LIMK1.
The objective of present research was to ascertain the function and mechanism of CBLB on apoptosis and ERS in LPS-induced type II alveolar epithelial cells (AECIIs). We proposed the hypothesis that m6A modification-mediated CBLB downregulation blocked the ubiquitination degradation of LIMK1, ultimately promoting ERS in LPS-induced AECIIs. An investigation into this mechanism has the potential to yield novel insights to develop clinical therapies for ALI.
Material and methods
Cell culture and treatment
The AECIIs (CP-H209, Procell Co. Ltd, Wuhan, China) were cultured in a humidity-controlled incubator at 37°C with 5% CO2 using DMEM (11965092, Gibco, NY, USA) containing 10% FBS. For LPS treatment, AECIIs were inoculated in 6-well plates (10,000 cells per well) and cultured overnight. Then the AECIIs were treated with LPS (10 µg/ml, SMB00610, Sigma-Aldrich, MO, USA) for 24 h to simulate ALI in vitro [18].
Cell transfection
Overexpression plasmids of CBLB (oe-CBLB) and LIMK1 (oe-LIMK1), shRNA targeting METTL14 (sh-METTL14) and CBLB (sh-CBLB), and control shRNA (sh-NC) and oe-NC were purchased from GenePharma (Shanghai, China). AECIIs were inoculated into 6-well plates and cultured for 24 h. When cell fusion was approximately 50%, plasmids were added to the cells concurrently with Lipofectamine 2000 (11668027, Thermo Fisher Scientific, MA, USA) for transfection. Following a 48-hour transfection period, the cells were collected. The efficacy of overexpression or knockdown was measured using RT-qPCR.
Cell Counting Kit-8 (CCK-8)
AECIIs were inoculated in 96-well plates (100 µl per well, containing approximately 2000 cells) and cultured in a 5% CO2 cell culture incubator at 37°C. After corresponding treatment, 10 µl of CCK-8 solution was added to each well and incubated in a 5% CO2 incubator at 37°C for 1 h. The absorbance at 450 nm was measured using a Microplate Reader as described in the Cell Counting Kit-8 (C0037, Beyotime, Shanghai, China).
Reactive oxygen species (ROS) assay
This experiment was conducted in strict accordance with the protocol outlined in the Reactive Oxygen Species Assay kit (S0033S, Beyotime). AECIIs were cultured in 6-well plates and subjected to the designated treatments. 1 ml of the 2’, 7’-dichlorodihydrofluorescein diacetate (DCFH-DA) probe, diluted in serum-free medium, was added to the AECIIs. The cells were incubated for 30 min at 37oC. Next, the cells were washed three times with a serum-free medium. A fluorescence microscope (Olympus, Tokyo, Japan) was then used to acquire the images.
TUNEL staining
This experiment was carried out as instructed in the Colorimetric TUNEL Apoptosis Assay Kit (C1091, Beyotime). AECIIs were fixed using 4% paraformaldehyde for 30 min, treated with 0.3% Triton X-100 for 5 min, and incubated with 0.3% hydrogen peroxide solution for 20 min. Subsequently, an appropriate amount of prepared biotin labeling solution was added to the above cells and reacted at 37oC for 1 h. At the completion of the reaction, the process was terminated with the reaction termination solution. The cells were incubated with the optimized concentration of streptavidin-horseradish peroxidase (HRP) working solution for 30 min, followed by treatment with diaminobenzidine (DAB) coloring solution for 10 min, then stained with hematoxylin for nuclear staining. Cells were washed with PBS after each step. The cells were meticulously examined under a microscope, and images were captured for further analysis.
Immunofluorescence (IF)
AECIIs were immobilized using 4% paraformaldehyde and then permeabilized using 0.1% Triton X-100 for 10 min. Next, the diluted GRP78 (ab21685, 1: 1000, Abcam, Cambridge, UK) primary antibody was added and incubated at 4oC overnight. After the primary antibody incubation, the cells were then rinsed three times (5 min each) with PBS, and incubated with biotin-labeled fluorescent secondary antibody protected from light for 30 min. Then, the cells were incubated with 4’,6-diamidino-2’-phenylindole (DAPI) protected from light for a period of 10 min. Fluorescence microscopy (Olympus, Tokyo, Japan) was employed to capture images.
Immunoprecipitation (IP)
The appropriate amount of IP lysis buffer was added to the collected cells for lysis. The supernatant of the sample was obtained through centrifugation. A portion of the supernatant was incubated with the appropriate amount of LIMK1 (19699-1-AP, 1: 400, Proteintech, Wuhan, China) antibody at 4oC overnight. Following incubation, protein A/G beads were added to the lysate for precipitation reaction. After the reaction, the beads were collected and the loading buffer was added to the beads. Western blotting was carried out to assess the level of ubiquitination in the samples.
Co-immunoprecipitation (Co-IP)
This protocol was conducted as described in the Co-IP kit (88804, Thermo Fisher Scientific). An appropriate amount of cell lysis buffer containing protease inhibitors was added to lyse AECIIs, and the supernatant was obtained by centrifugation after lysis. At the same time, part of the above cell lysate was retained for western blot, and then anti-CBLB (12781-1-AP, Proteintech) or anti-Immunoglobulin G (IgG, ab172730, Abcam) antibody was added to the remaining lysate and incubated overnight at 4oC. The above lysate was immunoprecipitated with Protein A/G beads. Then the beads were obtained through centrifugation. After washing with IP lysis buffer, the levels of LIMK1 and CBLB in the samples were analyzed via western blot.
Methylated RNA immunoprecipitation (MeRIP)
This experiment was conducted in strict accordance with the instructions of the MeRIP kit (17-10499, Millipore, MA, USA). After extraction of total cellular RNA, 450 ml of IP buffer and 2 µl of RNase inhibitor were added to the extracted RNA samples, and the RNA was fragmented using an ultrasonic crusher. Subsequently, 5 µg of anti- IgG (AC005, ABclonal, Wuhan, China) or anti-m6A (A19841, ABclonal) antibody was added to the samples, and the samples were incubated at 4°C for 2 h. Pre-treated Protein A/G beads were added and incubated at 4oC for 1 h. At the end of the incubation, the beads were placed in a magnetic rack and the supernatants were removed. The RNA was eluted after washing the beads three times, and RT-qPCR was used to ascertain the level of CBLB mRNA.
RNA immunoprecipitation (RIP)
The procedure was conducted in strict accordance with the directions of the RIP kit (17-704, Millipore). The mixed RIP lysis buffer containing 400 µl of RIP lysis buffer, 4 µl of protease inhibitor, and 1 µl of RNase inhibitor was added to the collected cell precipitates for lysis, and the supernatant was obtained by centrifugation after lysis. Appropriate amounts of anti-METTL14 (26158-1-AP, 1: 50, Proteintech), anti-METTL3 (15073-1-AP, 1: 50, Proteintech), anti-Wilms’ tumor 1-associating protein (WTAP, 60188-1-Ig, 1: 50, Proteintech), or anti-IgG (30000-0-AP, 1: 50, Proteintech) antibody were added to the pre-treated Protein A/G beads, and incubated at room temperature for 1 h. Then, the beads were placed in a magnetic rack, and the supernatants were discarded. Subsequently, the above cell lysates were added to the beads for incubation at 4oC overnight. Next, the beads were placed in a magnetic rack, and the supernatants were removed. The RNA obtained from the beads was subjected to RT-qPCR analysis to determine the levels of CBLB mRNA.
Actinomycin D assay
The AECIIs were inoculated into 6-well plates for normal culture. Then, the culture medium was replaced with a complete medium comprising 5 µg/ml actinomycin D (HY-17559, MedChemExpress, NJ, USA). Cells were collected at 0, 2, 4, 6, and 8 hours after actinomycin D treatment. RT-qPCR was used to quantitatively assess the CBLB mRNA level. The stability of CBLB mRNA was assessed through normalizing the expression of CBLB in the 0-hour group and calculating the ratio of the expression of CBLB in the remaining groups to the expression of CBLB in the 0-hour group.
Quantitative real-time PCR (RT-qPCR)
The AECIIs were lysed with Trizol (15596026CN, Invitrogen, CA, USA). The total RNA extraction process was carried out in strict accordance with the protocol outlined in the EZ-10 Total RNA Extraction Kit (B610583, Sangon Biotech, Shanghai, China) instructions. Then, the RNA was reverse transcribed to cDNA and amplified in accordance with the instructions outlined in PrimeScript RT-PCR Kit (RR014, Takara, Kyoto, Japan). The real-time qPCR protocol was executed with an ABI 7500 PCR system (Applied Biosystems, CA, USA). The relative target gene levels normalized to β-actin were determined with the 2-ΔΔCT method. Table 1 shows the primer sequences.
Western blot
The AECIIs were collected and lysed for 30 min at 4oC with RIPA Lysis Buffer (89901, Thermo Fisher Scientific). Subsequently, the protein content of each sample was detected with a BCA Protein Assay Kit (P0012, Beyotime). SDS-PAGE gels were applied to separate the protein samples, and the separated proteins were transferred to PVDF (ISEQ00010, Millipore). After blocking the membranes with 5% skim milk for 1 h, they were incubated overnight at 4°C with the following primary antibodies: CBLB (12781-1-AP, 1 : 1000, Proteintech), GRP78 (ab21685, 1 µg/ml, Abcam), CHOP (#2895, 1: 1000, Cell Signaling Technology), Protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK, #3192, 1 : 1000, Cell Signaling Technology), p-PERK (29546-1-AP, 1 : 1000, Proteintech), LIMK1 (ab95186, 1: 2000, Abcam), METTL14 (26158-1-AP, 1 : 1000, Proteintech), and β-actin (ab8226, 1 µg/ml, Abcam). Then, the membranes were incubated with the specific secondary antibodies (1: 10000, ab7090, ab6728, Abcam) for 1h at ambient temperature. The membranes were visualized and images captured using a GEL imaging system (Bio-Rad, CA, USA).
Statistical analysis
Analysis of the data was conducted using GraphPad Prism 9 (GraphPad Software, CA, USA). Measurements that conformed to a normal distribution were expressed as mean ± standard deviation. Comparisons between two groups were carried out using the t-test. Comparisons between multiple groups were assessed by one-way ANOVA followed by Tukey’s multiple comparison test. A value of p < 0.05 was considered statistically significant.
Results
CBLB overexpression inhibited apoptosis and ERS in LPS-induced AECIIs
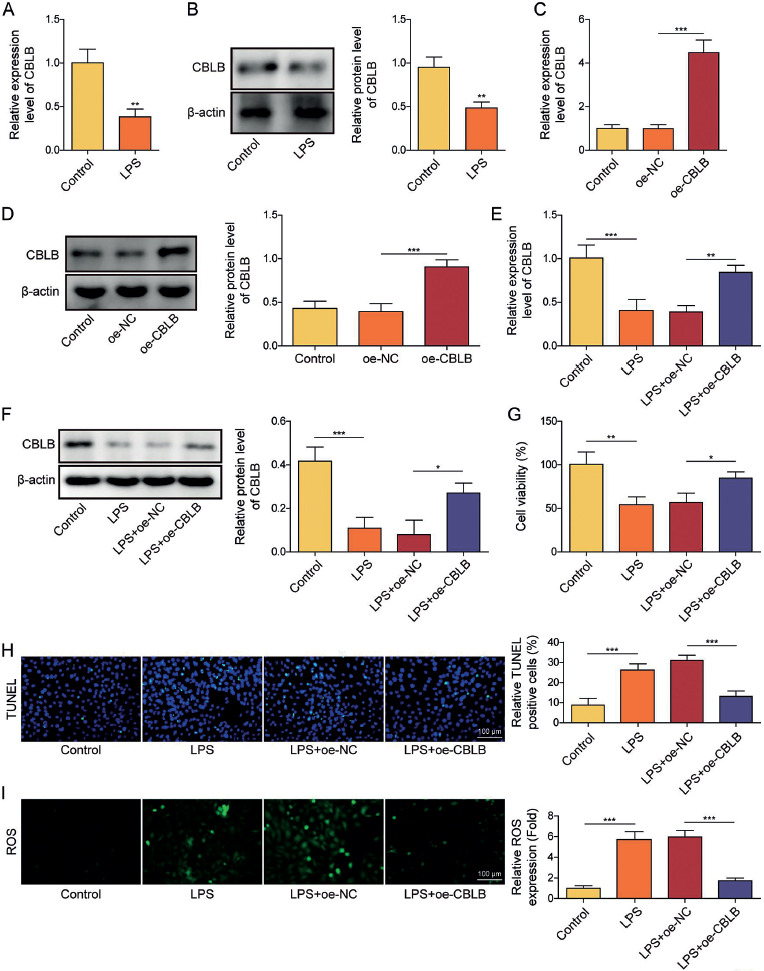
Firstly, AECIIs were treated with LPS for 24 h to simulate ALI in vitro. It was found that both mRNA and protein levels of CBLB were significantly downregulated in AECIIs after treatment with LPS (Fig. 1A, B). Subsequently, we transfected the oe-CBLB plasmid in AECIIs and the results revealed that transfection of oe-CBLB plasmid markedly augmented the levels of CBLB mRNA and protein in AECIIs (Fig. 1C, D). After overexpression of CBLB in LPS-induced AECIIs, the LPS-induced downregulation of CBLB was significantly reversed (Fig. 1E, F). The results further showed that LPS treatment weakened the viability of AECIIs and promoted apoptosis, which was counteracted by CBLB overexpression (Fig. 1G, H). Additionally, upregulation of ROS caused by LPS was inhibited by CBLB overexpression (Fig. 1I). Also, the ERS-related protein (including p-PERK, CHOP, and GRP78) levels were increased by LPS, and this effect was abolished by overexpression of CBLB (Fig. 1J, K). These results suggest that overexpression of CBLB suppressed apoptosis and ERS in LPS-induced AECIIs.
Fig. 1
Cont. J) Western blot was applied to assay protein levels of GRP78, CHOP, p-PERK, and PERK in AECIIs.K) Protein level of GRP78 in AECIIs was examined by IF (scale bar = 100 μm). Red fluorescence indicated GRP78 positivity, while blue fluorescence indicated DAPI positivity. AECIIs in E-K were treated with LPS and transfected with oe-CBLB plasmids. One-way ANOVA was performed for statistical analysis (C-K). n = 3, *p < 0.05, **p < 0.01,***p < 0.001
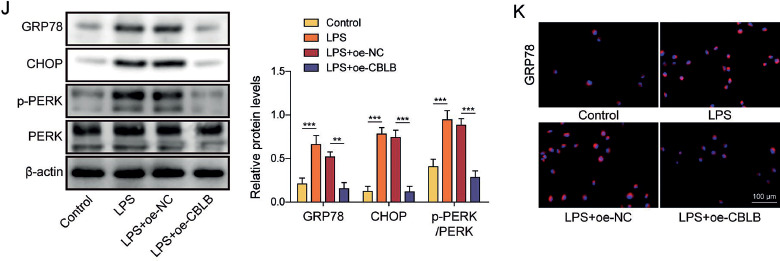
CBLB promoted the degradation of LIMK1 via ubiquitination in AECIIs
Then we investigated the molecular mechanism by which overexpression of CBLB inhibited apoptosis and ERS in LPS-induced AECIIs. First, LIMK1, a serine/threonine kinase, was highly expressed in LPS-induced AECIIs (Fig. 2A). Then, we found that LIMK1 might be a potential ubiquitination substrate for CBLB using the UbiBrowser database (Fig. 2B). Additionally, the ubiquitination level of LIMK1 was examined using IP assay, and it was found that LPS treatment reduced the ubiquitination level of LIMK1 in AECIIs (Fig. 2C). Furthermore, the results of Co-IP showed that the anti-CBLB antibody markedly enriched LIMK1 protein compared to anti-IgG antibody, indicating the interaction of LIMK1 and CBLB (Fig. 2D). CBLB overexpression did not alter LIMK1 mRNA levels but downregulated LIMK1 protein levels (Fig. 2E, F); it augmented the ubiquitination of LIMK1 and weakened the protein stability of LIMK1 in AECIIs (Fig. 2G, H). In addition, CBLB overexpression in LPS-stimulated AECIIs significantly counteracted the upregulatory effect of LPS on LIMK1 protein levels (Fig. 2I). Overall, it was found that CBLB decreased the protein stability of LIMK1 through ubiquitination in AECIIs.
Fig. 2
CBLB promoted degradation of LIMK1 via ubiquitination in AECIIs. A) Protein level of LIMK1 in AECIIs was detected by western blot. AECIIs were treated with LPS. B) UbiBrowser database was employed to predict LIMK1 as a substrate for ubiquitination by CBLB. C) Ubiquitination level of LIMK1 in AECIIs was assayed via IP combined with western blot. AECIIs in C were treated with LPS. D) Binding of CBLB and LIMK1 in AECIIs was verified by Co-IP combined with western blot. E) Level of LIMK1 mRNA was measured by RT-qPCR. F) Western blot was used to detect the level of LIMK1 protein. G) IP combined with western blot was used to evaluate the ubiquitination level of LIMK1 in AECIIs. AECIIs in E-G were transfected with oe-CBLB plasmids. H) Level of LIMK1 protein in AECIIs treated for different times with CHX (0, 30, 60, and 90 min) was assessed by western blot. AECIIs in H were treated with CHX and transfected with oe-CBLB plasmids. I) Level of LIMK1 protein in AECIIs was assayed by western blot. AECIIs in I were treated with LPS and transfected with oe-CBLB plasmids. Student’s t-test was performed for statistical analysis (A-H). One-way ANOVA was performed for statistical analysis (I). n = 3, **p < 0.01, ***p < 0.001
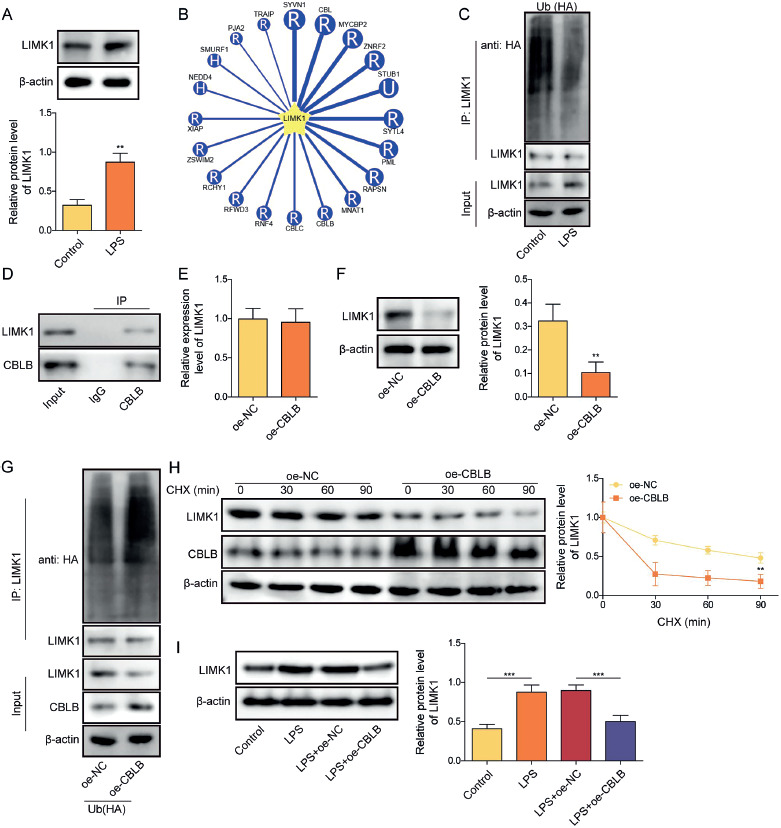
Overexpression of LIMK1 reversed the effects of CBLB overexpression on apoptosis and ERS in LPS-induced AECIIs
Consequently, we further investigated whether the overexpression of CBLB inhibited apoptosis and ERS in LPS-treated AECIIs by regulating LIMK1. Firstly, we transfected the oe-LIMK1 plasmids in AECIIs, and the results showed that transfection of oe-LIMK1 plasmid markedly elevated the levels of LIMK1 mRNA and protein in AECIIs (Fig. 3A, B). After that, we overexpressed CBLB or concurrently overexpressed LIMK1 in LPS-induced AECIIs. Our findings revealed that the downregulation effect of CBLB upregulation on LIMK1 was significantly abolished by LIMK1 overexpression (Fig. 3C). The upregulated CBLB significantly enhanced cell viability, inhibited apoptosis, decreased the level of ROS, and inhibited the levels of p-PERK, CHOP, and GRP78 in LPS-induced AECIIs; all the above effects of CBLB overexpression were reversed by oe-LIMK1 (Fig. 3D-H). These results demonstrated that overexpression of CBLB attenuated apoptosis and ERS in LPS-induced AECIIs by suppressing LIMK1 expression.
Fig. 3
Overexpression of LIMK1 reversed the effects of CBLB overexpression on apoptosis and ERS in LPS-induced AECIIs. A, B) Transfection efficiency of oe-LIMK1 was assayed using RT-qPCR and western blot. C) Protein level of LIMK1 in AECIIs were evaluated via western blot. D) CCK-8 was applied to examine cell viability in each group.E) Apoptosis of AECIIs was assayed via TUNEL staining (scale bar = 100 μm). F) ROS level was observed by fluorescence microscopy after addition of DHE fluorescent probe to each group of AECIIs (scale bar = 100 μm). G) Western blot was used to detect protein levels of GRP78, CHOP, p-PERK, and PERK in AECIIs. H) Protein level of GRP78 in AECIIs was measured by IF (scale bar = 100 μm). Red fluorescence represented GRP78 positivity and blue fluorescence represented DAPI positivity. AECIIs in C-H were treated with LPS and transfected with oe-CBLB and oe-LIMK1 plasmids. One-way ANOVA was performed for statistical analysis. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001
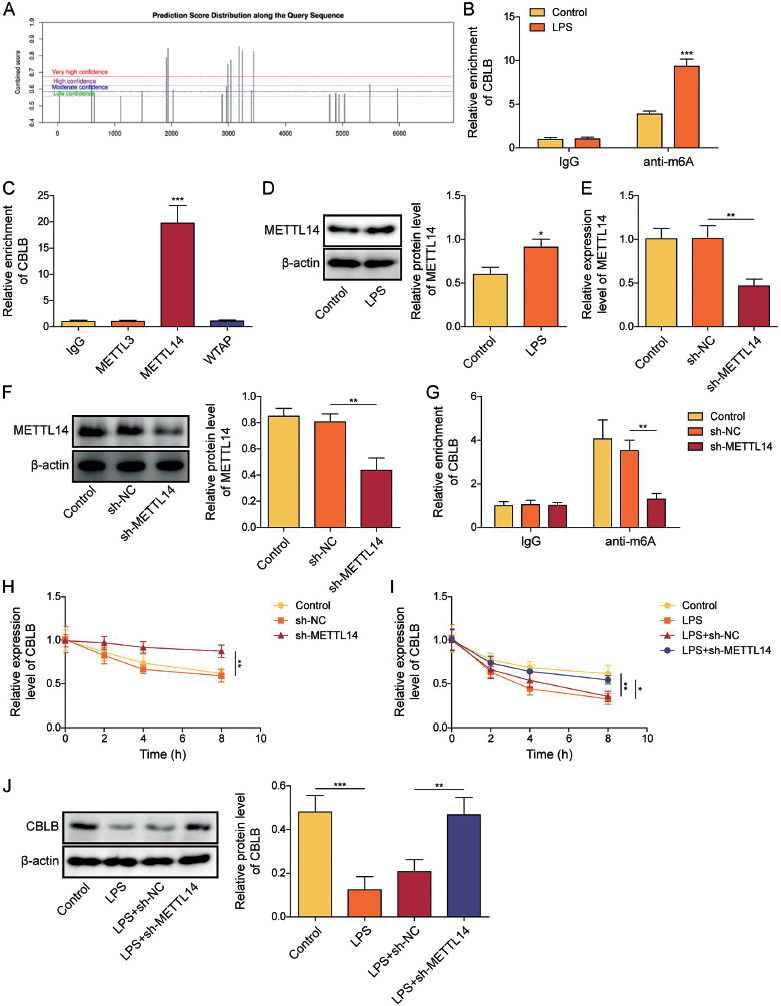
METTL14 impaired the stability of CBLB mRNA by increasing its m6A modification
Then we investigated the mechanism underlying the decreased expression of CBLB in LPS-induced AECIIs. We found that the CBLB mRNA contained multiple m6A modification sites using the SRAMP database (Fig. 4A). The MeRIP experiment showed that the m6A modification level of CBLB mRNA was augmented in AECIIs after LPS treatment (Fig. 4B). Therefore, we further explored the methyltransferase involved in the m6A modification of CBLB by RIP experiments and found that CBLB RNA bound exclusively to methyltransferase 14 (METTL14) (Fig. 4C). LPS treatment resulted in elevated protein levels of METTL14 in AECIIs (Fig. 4D). Subsequently, we transfected shRNA targeting METTL14 in AECIIs and verified the knockdown efficacy of sh-METTL14 through RT-qPCR and western blot (Fig. 4E, F). The findings revealed that the level of m6A modification of CBLB in AECIIs was reduced (Fig. 4G) and the stability of CBLB mRNA (Fig. 4H) was increased after knockdown of METTL14. Additionally, the stability of CBLB mRNA was reduced in AECIIs under LPS treatment. Nevertheless, the silencing of METTL14 enhanced the stability and expression of CBLB mRNA in LPS-induced AECIIs (Fig. 4I, J). These results indicated that METTL14 attenuated the stability of CBLB mRNA by elevating the m6A modification of CBLB mRNA in LPS-induced AECIIs.
Fig. 4
METTL14 reduced the stability of CBLB mRNA in a m6A-dependent manner. A) SRAMP database was used to predict the m6A modification site of CBLB. B) MeRIP was used to identify the m6A modification level of CBLB in AECIIs after LPS treatment. C) Interactions between CBLB mRNA and m6A methyltransferases (including METTL3, METTL14, and WTAP) in AECIIs were verified by RIP. D) Protein level of METTL14 in AECIIs was assayed via western blot after LPS treatment. E, F) Knockdown efficiency of sh-METTL14 was assayed using RT-qPCR and western blot. G) m6A modification level of CBLB in AECIIs was examined by MeRIP. AECIIs in E-G were transfected with shRNA targeting METTL14. H) RT-qPCR was employed to detect the level of CBLB mRNA. AECIIs in H were treated with actinomycin D and transfected with shRNA targeting METTL14. I) CBLB mRNA level was assessed by RT-qPCR. J) Western blot was used to analyze the level of CBLB protein. AECIIs in I-J were treated with LPS and transfected with shRNA targeting METTL14, followed by actinomycin D treatment. Student’s t-test was performed for statistical analysis (B, D). One-way ANOVA was performed for statistical analysis (C, E-J). n = 3, *p < 0.05, **p < 0.01, ***p < 0.001
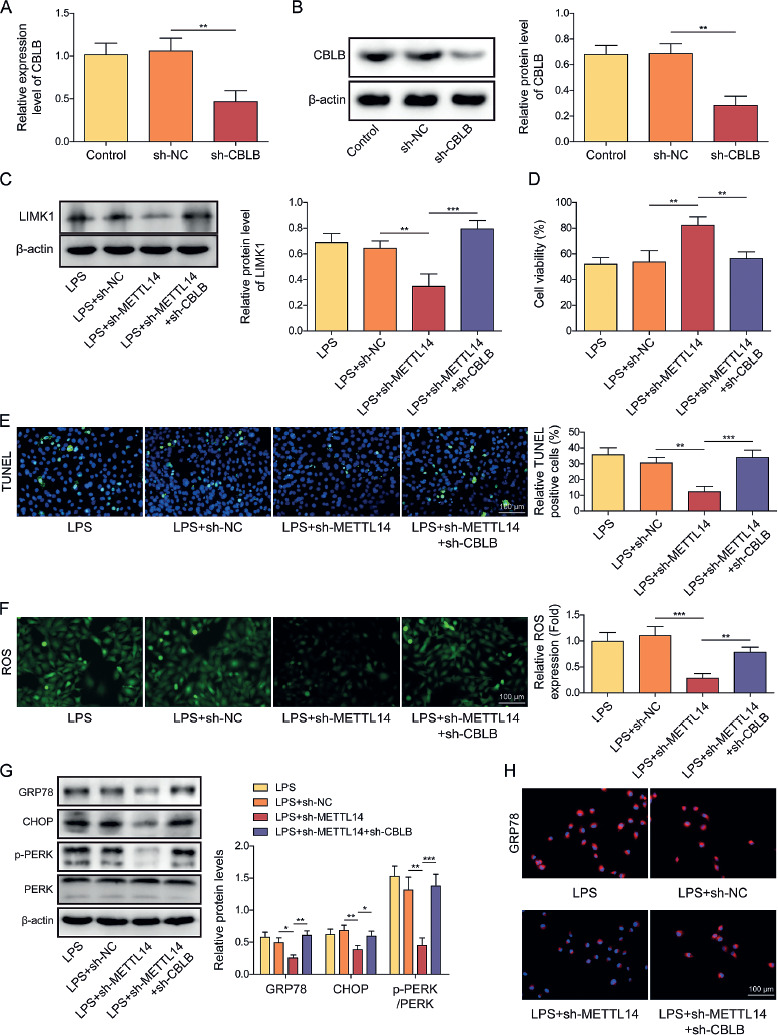
Knockdown of CBLB overturned the effects of METTL14 knockdown on LPS-stimulated LIMK1 expression levels, apoptosis, and ERS
To investigate whether the regulatory action of CBLB on apoptosis and ERS in LPS-induced AECIIs was affected by METTL14, we knocked down METTL14 or simultaneously silenced CBLB in LPS-induced AECIIs. Firstly, the transfection of sh-CBLB significantly decreased the CBLB level in LPS-induced AECIIs (Fig. 5A, B). The findings showed that the suppression of METTL14 decreased the protein level of LIMK1 in LPS-induced AECIIs, which was abolished by CBLB knockdown (Fig. 5C). Furthermore, the knockdown of METTL14 was observed to enhance the viability, inhibit apoptosis, decrease the level of ROS, and inhibit the levels of p-PERK, CHOP, and GRP78 in LPS treated AECIIs, whereas these effects were reversed by the simultaneous CBLB inhibition (Fig. 5D-H). These results indicated that METTL14 triggered apoptosis and ERS in LPS-induced AECIIs by decreasing CBLB.
Fig. 5
Knockdown of CBLB reversed the effects of METTL14 knockdown on LPS-induced LIMK1 expression levels, apoptosis, and ERS. A, B) Knockdown efficiency of sh-CBLB was assayed using RT-qPCR and western blot. C) Protein level of LIMK1 in AECIIs was evaluated by western blot. D) CCK-8 was applied to detect the viability of AECIIs in each group. E) TUNEL staining was used to evaluate the level of apoptosis in AECIIs (scale bar = 100 μm). F) ROS level was observed using fluorescence microscopy after addition of DHE fluorescent probe to each group of AECIIs (scale bar = 100 μm). G) Western blot was used to analyze protein levels of GRP78, CHOP, p-PERK, and PERK in AECIIs. H) Protein level of GRP78 in AECIIs was evaluated via IF (scale bar = 100 μm). Red fluorescence indicated GRP78 positivity, while blue fluorescence indicated DAPI positivity. AECIIs in C-H were treated with LPS and transfected with shRNA targeting METTL14 and CBLB. One-way ANOVA was performed for statistical analysis. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
The regeneration and proliferation of AECIIs can effectively prevent the onset of ALI [19]. Previous research demonstrated that the attenuation of ERS effectively mitigated the apoptosis of AECIIs in the presence of an unfavorable stimulus [5]. The current findings suggested that the enhancement of CBLB inhibited apoptosis and ERS in LPS-induced AECIIs by promoting the degradation of LIMK1. Secondly, we found that METTL14 reduced the stability of CBLB mRNA through enhanced m6A modification of CBLB mRNA. CBLB may be a key factor affecting ERS in alveolar epithelial cells, and specific intervention of CBLB and its upstream and downstream regulatory factors may be an effective way to improve ALI.
When stimulated by some exogenous or endogenous factors, the accumulation of misfolded or unfolded proteins triggers ERS [20]. This is followed by the ectopic translocation of the molecular chaperone GRP78/BiP within the ER, which causes the phosphorylation of transmembrane proteins (including inositol-requiring enzyme 1α (IRE1 α), PERK, and activating transcription factor 6 (ATF6)) [21]. Additionally, phosphorylated IRF1 α promotes JNK-mediated signaling, which in turn activates apoptosis [22]. In the event of excessive ERS, PERK-mediated activation of ATF4 initiates CHOP transcription to enhance apoptosis [21]. In instances of severe acute injury, the repair and regeneration mechanisms of AECIIs are unable to compensate for the damage, leading to the development of ALI [23]. ERS plays an important role in AEC injury. For instance, the administration of exogenous arachidonic acid metabolites was proven to attenuate the senescence of AECs by inhibiting ERS [24]. Furthermore, it was observed that the progression of bleomycin-induced lung inflammation and pulmonary fibrosis in rats could be effectively ameliorated by suppressing ERS-mediated apoptosis in AECIIs [25]. Herein, our study emphasized again that LPS-induced AECII injury was associated with increased ERS levels, and effective intervention of ERS might be an important way to reduce AECIIs damage and thus ameliorate ALI.
CBLB is a crucial E3 ubiquitin ligase that facilitates the ubiquitination of downstream proteins [7]. It was illustrated that CBLB impeded the development of T follicular helper cells via enhancing the ubiquitination-mediated degradation of inducible synergistic co-stimulation molecules, consequently hindering the progression of systemic lupus erythematosus [26]. Furthermore, CBLB was identified as a highly promising small molecule immunotherapy target due to its function in regulating the maturation and migration of a diverse range of immunosuppressive cells [27]. In the context of acute myocardial infarction, the overexpression of CBLB significantly diminished inflammatory cell infiltration and collagen fiber formation in the region of myocardial infarction [28]. The deficiency of CBLB was observed to enhance the LPS-induced production of inflammatory mediators and reactive oxygen species in bone marrow-derived macrophages, thereby accelerating the progression of atherosclerosis [29]. Hence, it was concluded that CBLB had an inhibitory effect on the inflammatory response in a number of different diseases. Importantly, it was pointed out that CBLB played a significant role in the ERS [8]. The findings of the current study indicated that the CBLB expression was markedly diminished in LPS-induced AECIIs. Additionally, this is the first study reporting that augmenting CBLB expression mitigated apoptosis and ERS in LPS-treated AECIIs.
LIMK1, as an essential protein kinase, is implicated in multiple biological procedures, including the cell cycle and cell migration, by regulating cytoskeletal dynamics [14]. LIMK1 has been demonstrated to positively regulate the inflammatory response in multiple diseases. The repression of LIMK1 expression minimized the levels of inflammatory factors and apoptosis-related markers in rats with heart failure [30]. In particular, recent observations indicated that in LPS-induced ALI models, inflammatory cell infiltration could be hindered and lung tissue injury could be attenuated by inhibiting the transduction of LIMK1-related signaling pathways [16]. We also observed that LIMK1 expression was elevated in LPS-treated AECIIs. Furthermore, this study elucidated the role for CBLB as an E3 ubiquitin ligase, facilitating the degradation of LIMK1 in LPS-induced AECIIs. It was previously found that overexpression of LIMK1 elevated the levels of ERS-related proteins (including p-PERK, CHOP, and BIP) to regulate the occurrence of ERS and consequently contribute to 5-FU resistance in colorectal cancer cells [17]. The findings of this study demonstrated that overexpression of CBLB inhibited LPS-induced ERS in AECIIs by degrading LIMK1. Given the kinase activity of LIMK1, the majority of existing studies concentrated on the observation that LIMK1 mediated the phosphorylation-dependent inactivation of the actin-binding factor cofilin to regulate actin polymerization [31]. Chen et al. proposed that eIF2 α in the PERK pathway might serve as a direct downstream target of LIMK1. They further suggested that LIMK1 phosphorylated and activated eIF2α to increase the expression of ATF4 and regulate the ERS response pathway [17]. The above suggests that the role of LIMK1 in promoting ERS in ALI may be related to the kinase activity of LIMK1. This hypothesis will be further investigated in subsequent research.
M6A modifications had the capacity to modulate all steps of the RNA cycle, including RNA processing, nuclear export, degradation, and translation [11]. The regulation of m6A modification of specific genes can either accelerate or slow down the progression of ALI. For example, as shown in studies conducted by other researchers on sepsis-associated ALI, METTL3 mediated m6A modification of HIF-1 α to strengthen the mRNA stability of HIF-1α, which exacerbated ferroptosis in AECs [13]. METTL14, another important methyltransferase mediating m6A modification, was reported to aggravate ALI by augmenting the stability of nucleotide-binding oligomerization domain-like receptor protein 3 mRNA, which depended on the recognition of m6A modifications by insulin like growth factor 2 mRNA binding protein 2 [32]. Moreover, METTL14 was reported to promote the ERS response and apoptosis in an m6A-dependent manner [33]. In the current study, METTL14 enhanced the m6A modification of CBLB mRNA in LPS-induced AECIIs. Furthermore, the repressive effect of METTL14 knockdown on LPS-induced apoptosis and ERS in AECIIs was counteracted by CBLB silencing. Accordingly, the present study clarified that METTL14 augmented apoptosis and ERS in LPS-induced AECIIs by raising the m6A methylation modification of CBLB mRNA to negatively regulate the mRNA stability and expression of CBLB. It is well established that the outcome of m6A methylation-modified mRNA is dependent on the action of the m6A reader. For example, the m6A reader YTHDF2 is widely reported to bind to m6A-modified RNAs and promote the degradation of target RNAs [34]. Conversely, various members of the IGF2BPs family were found to maintain the stability of target RNAs by recognizing m6A modifications [35]. Therefore, it was worthwhile to carry out further exploration in subsequent studies to ascertain which m6A-reading protein bound to m6A-modified CBLB in ALI.
In summary, the current study illustrated that METTL14 modified CBLB mRNA with m6A methylation to undermine the mRNA stability of CBLB, which reduced the expression of CBLB in LPS-induced AECIIs. Downregulated CBLB impeded ubiquitination and degradation of LIMK1 to promote ERS in LPS-induced AECIIs. The targeted inhibition of METTL14 or promotion CBLB is expected to be a novel approach to the management of ALI. Lastly, this study is merely a preliminary validation of our hypothesis at the cellular level, which needs to be further confirmed by animal and clinical experiments.