Introduction
Lung macrophages are critical players in the immune system, serving as the first line of defense in the lungs, a key organ continuously exposed to external pathogens and environmental particles. These tissue-resident cells are strategically positioned to monitor and respond to foreign invaders, ensuring both the clearance of pathogens and the maintenance of tissue homeostasis. Their role extends beyond simple pathogen elimination; they are also involved in regulating inflammation, presenting antigens to adaptive immune cells, and orchestrating the repair and regeneration of lung tissue following injury.
In recent years, the understanding of lung macrophage biology has greatly expanded. Technological advancements such as single-cell transcriptomics, CRISPR gene editing, and proteomics have revealed the heterogeneity, functional plasticity, and developmental origins of lung macrophages. These insights have profound implications for understanding how macrophages contribute to both lung health and disease, particularly in such conditions as acute respiratory distress syndrome (ARDS), asthma, chronic obstructive pulmonary disease (COPD), and respiratory infections, including influenza and tuberculosis (TB).
Despite these advancements, key challenges remain. The molecular mechanisms governing macrophage function are still unclear and require deeper exploration. Understanding how macrophages interact with other immune cells and their role in disease progression remains critical for developing targeted therapies. Additionally, how macrophage dysfunction contributes to the pathogenesis of various pulmonary diseases needs further study to uncover new therapeutic targets and improve patient outcomes. This review aims to provide a comprehensive overview of the current understanding of lung macrophage biology, their roles in various pulmonary diseases, and the potential therapeutic approaches that target macrophage-mediated pathways to improve the prognosis of patients with lung diseases. By delving into these issues, we can better understand the key role of lung macrophages in maintaining pulmonary health and fighting diseases, and provide direction for future research and treatment.
Lung macrophages: characteristics and functions
Development and differentiation
The lungs, as the core organ of the human respiratory system, not only dominate the basic life process of gas exchange, but also deeply participate in various physiological activities, including immune regulation, pulmonary circulation maintenance, and hematopoietic function [1]. Frequent environmental exposure makes the lungs a major entry point for pathogens. Tissue-resident macrophages play essential roles in host defense, tissue repair, and maintaining homeostasis. Under homeostatic conditions, macrophages are distributed across the lung: the luminal side of the alveolar lumen (the inner surface of the alveolar space, directly exposed to inhaled air), the interalveolar stroma, the submucosa of the fine bronchioles, and the vascular epithelium. Their classification depends on their location within the lung [2, 3]. Among the various subtypes, alveolar macrophages (AMs), located in the alveoli and airway lumens, are of the most concern. These cells originate from embryonic precursors and are primarily involved in immune surveillance, clearing foreign bodies, and balancing pro- and anti-inflammatory responses. Additionally, interstitial macrophages (IMs), residing in the lung interstitium, derive from both bone marrow- derived macrophages (BMDMs) and embryonic progenitors. IMs play key roles in immune regulation, tissue repair, and interaction with other immune cells [4].
Alveolar macrophages
Alveolar macrophages (AMs) are a dominant population of innate immune cells in the distal lung parenchyma (the lung region beyond the terminal bronchioles, primarily consisting of alveoli and associated structures), residing along the luminal surface of the alveolar space [5]. Embedded amidst a microenvironment comprising type I and II alveolar epithelial cells (AECs), capillary endothelial cells, and alveolar interstitial fibroblasts, this niche fosters a cytokine-laden microcosm that nurtures the proliferation and function of AMs. Proximal to the mucosal surface, AMs play an indispensable role in maintaining airway homeostasis [6, 7]. They are abundant in the lungs, with the upper lobe of the human lung containing 1.5 × 109 AMs, most of which are confined to the lung’s diffusion area, while a few are limited to the conducting small airways [8]. The development and maturation of AMs, as well as their self-maintenance and renewal, require granulocyte-macrophage colony-stimulating factor (GM-CSF) and transforming growth factor β (TGF-β) secreted by alveolar type II epithelial cells [9-11]. In stable physiological states, AMs primarily derive from embryonic precursors, exhibiting an autonomous maintenance mechanism that is decoupled from the circulating monocyte pool. In the specific context of mice, these AMs originate from fetal monocytes that, guided by the orchestration of GM-CSF, TGF-β, and PPARγ (peroxisome proliferator activated receptor γ) signaling pathways, migrate to the lungs postnatally and subsequently undergo differentiation into fully mature AMs.
Lung macrophages exhibit remarkable plasticity, shifting between M1 and M2 phenotypes depending on environmental signals [12]. The M1 phenotype is typically activated by Th1 cytokines such as interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α), and they release pro-inflammatory cytokines including interleukin (IL)-1, IL-6, IL-12, IL-23, and TNF-α, as well as nitric oxide, which are crucial for pathogen elimination. Conversely, the M2 phenotype, induced by Th2 cytokines such as IL-4 and IL-13, is distinguished by the production of anti-inflammatory cytokines, including IL-10 and TGF-β, which are essential for mediating tissue repair, immune modulation, and the resolution of inflammation [13, 14]. Apart from the M1 and M2 types, other less common macrophage subtypes such as the M4, Mhem, and Mox phenotypes have been identified, each with distinct roles in various biological processes [13].
While defending against pathogenic invasion, M1 macrophages may simultaneously cause acute lung damage. On the other hand, M2 macrophages can be further differentiated into subtypes such as M2a, M2b, and M2c based on the inducing factors. The M2a subtype, typically induced by IL-4 or IL-13, primarily facilitates repair of damaged tissue. The M2b subtype is generated by immune complexes (IC) and toll-like receptor (TLR) or IL-1 receptor (IL-1R) ligands, participating in the processing of IC and modulation of inflammation. The M2c subtype, induced by IL-10 and glucocorticoids (GC), possesses immunomodulatory and inhibitory effects, and also promotes tissue repair, which is beneficial for the healing of lung tissue damage [13, 15, 16].
Interstitial macrophages
Interstitial macrophages (IMs) originate from both BMDMs and embryonic progenitor cells [17]. Occupying the intricate realm between the lung epithelium and capillaries, they densely populate the lung interstitium [18]. These cells engage in intricate interactions with other immune sentinels, including lymphocytes and dendritic cells (DCs), within this microenvironment. Nonetheless, despite their pivotal role, the precise anatomical localization of IMs remains elusive in the majority of research endeavors, leaving open the possibility that they may be implicated in lung barrier immunity, alongside AMs, within the alveolar interstitium, submucosa, or even the perivascular epithelium. Research has found that AMs sorted from rat lungs are larger and have more pseudopodia; IMs are smaller than AMs, have smoother surfaces, and their nuclei are more irregular. It has been reported that IMs, compared to AMs, display nuclei with a relatively higher proportion of condensed chromatin. While heterochromatin is a universal characteristic of eukaryotic nuclei, this relative difference in chromatin condensation may reflect distinct functional states between these cell types [19]. Currently, the research landscape pertaining to IMs lags behind that of AMs, primarily owing to two pivotal factors. Firstly, AMs exhibit a distinct advantage in accessibility and identifiability, facilitating their retrieval from the lungs of both animal models and human patients through bronchoalveolar lavage (BAL). BAL offers a relatively straightforward approach to acquiring AMs. It typically yields a cell population dominated by AMs (> 90%), along with smaller proportions of lymphocytes, neutrophils, eosinophils, and occasionally epithelial cells, depending on the inflammatory or pathological state of the lung. The differential cell counts obtained from BAL fluid are valuable diagnostic indicators for various pulmonary conditions, including infections, interstitial lung diseases, and malignancies [20]. However, it is crucial to acknowledge that this technique primarily captures the “active” subset of AMs, potentially overlooking the “stationary” population attached to the alveolar epithelium, thus potentially introducing a bias in sample representation. In stark contrast, the investigation of IMs necessitates more invasive procedures, including lung resection in animal models and lung tissue biopsy or surgery in humans. Subsequently, these samples undergo intricate tissue dissociation and rigorous cell purification protocols to isolate IMs, posing significant technical hurdles that inadvertently skew research efforts towards AMs. Furthermore, IMs inherently represent a fleeting transitional phase of tissue infiltration by circulating monocytes, destined to differentiate into AMs within the airway lumen. This transient nature further complicates their study, adding another layer of complexity to the already challenging research landscape [21].
Despite technical limitations that have hindered the precise isolation and differentiation of IMs from monocytes and DCs, the increased utilization of scRNA-seq technology in recent years has provided novel insights into the identification and classification of IMs.
Recent research has identified that IMs in the lungs of rats can be divided into three distinct subsets based on the relative surface expression of CD11c and MHC-II: CD11clowMHC-IIlow (IM1), CD11clowMHC-IIhigh (IM2), and CD11cMHC-II+high (IM3) [22, 23]. Phenotypic analysis reveals that IM1 and IM2 express higher levels of CD206, Lyve-1, and CD169, while IM3 exhibits higher levels of CCR2 (chemokine receptor type 2, also known as CD192) and CD11c. Functionally, IM1 and IM2 appear to be more efficient than IM3 in certain respects, yet they exhibit lower phagocytic efficiency for latex microspheres or microbial bioparticles in vivo compared to AMs. However, in vitro experiments demonstrate similar phagocytic capabilities across all three populations [22]. This suggests a potential diversity within IMs under homeostatic conditions. When the lungs are exposed to endogenous or exogenous stress signals, such as after tissue injury or during inflammation or infection, the situation may become more complex. In these contexts, IMs may adjust their phenotypes and functions to meet the demands of the pulmonary tissue [23]. A comparison of the properties of AMs and IMs at steady state is shown in Table 1.
Associated markers for the different subpopulations
Different subpopulations of lung macrophages exhibit distinct phenotypic markers and functions under various physiological and pathological conditions. The activation of M1 macrophages within AMs occurs through signaling pathways involving IFN-γ, TNF, and TLRs [14]. The surface markers for these cells primarily include CD68, CD80, CD86, and CD32 [24]. Microbial products or pro-inflammatory cytokines induce the polarization of M1 macrophages, in which IFN-α can trigger specific gene expression profiles, including MHC-II, IL-12, inducible nitric oxide synthase (iNOS), and suppressor of cytokine signaling-1 (SOCS-1) [25]. In contrast, the activation of M2 macrophages is driven by cytokines such as IL-4, IL-10, and IL-13, with surface markers mainly comprising CD115, CD206, and CD163 [24]. It is important to note that due to the phenotypic continuum between M1 and M2 macrophages, intermediate macrophages can co-express some of these markers. IMs in the lung play a crucial role in maintaining pulmonary homeostasis, regulating inflammatory responses, and promoting tissue repair. Their marker expression and functional states are finely regulated by the local microenvironment, including markers such as CD11b, CD14, CD64, and CD169 [19]. In addition to surface markers, several molecular markers have been identified as specific to M2 macrophages through immunohistochemistry and flow cytometry. These markers include scavenger receptor-A CD204, CD209 (also known as dendritic cell-specific intercellular adhesion molecule-1-3 grabbing non-integrin, DC-SIGN), and TREM-1 [26]. Single-cell omics studies have further revealed transcription factors associated with lung macrophages. For instance, IRF5 and STAT1 are highly expressed in M1 macrophages [27], while STAT6 and IRF4 are highly expressed in M2 macrophages [28]. The surface markers mentioned above are generally conserved across mammals; however, some markers, such as F4/80, are predominantly used in murine macrophages and may not be applicable to other mammalian species. A comparison of the properties of AMs and IMs at steady state is shown in Table 1.
Table 1
Properties of alveolar macrophages (AMs) and interstitial macrophages (IMs) in the steady state
Basic functions and immune defense
Lung macrophages are pivotal for maintaining pulmonary homeostasis and defending against pathogenic invasions. They rapidly identify and phagocytose pathogens that enter the lungs, such as bacteria, viruses, and fungi. Via pattern recognition receptors (PRRs) on their cell surface, they recognize pathogen-associated molecular patterns (PAMPs) and initiate phagocytosis, encapsulating pathogens within phagosomes for degradation. This process not only effectively reduces the number of pathogens in the lungs but also limits the spread of infection [29]. Furthermore, lung macrophages play a key role in immune surveillance by presenting antigens to T cells and initiating adaptive immune responses. They also promote tissue homeostasis and repair by secreting anti-inflammatory cytokines and growth factors [14].
Alveolar macrophages reside within the alveolar lumen, where gas exchange occurs across the alveolar-capillary membrane. They remove microbes, dead cells, and airborne particles via phagocytosis, which is crucial for maintaining airway patency and efficient oxygen exchange [4]. AMs serve as the first line of defense against bacterial and fungal pathogens, with their phagocytic activity increasing in response to infection. IMs are major producers of IL-10 and can inhibit LPS-induced DCs maturation and migration by secreting high levels of IL-10 [30-32]. IMs enhance their capacity to produce IL-10 during homeostasis and in response to microbial products. This characteristic suggests that IMs can exert regulatory functions [33]. The expression of IL-10 in human and murine IMs increases upon environmental immune stimulation, and although the signaling pathways that promote IL-10 expression in IMs are not well studied, current research has demonstrated that signaling through MyD88, a downstream adapter protein for TLRs and IL-1 receptors, is upstream of the pro-inflammatory transcription factor hypoxia-inducible factor-1α (Hif-1α) [34]. These features indicate that IMs effectively play a negative feedback role in inflammatory responses [21, 34].
Inflammation regulation and tissue repair
During infection or injury, lung macrophages detect PAMPs via TLR-4. This recognition process triggers a complex signaling cascade response that centrally involves multiple key signaling pathways such as NF-κB, TLR, interferon and ER-phagosome, which further activates and promotes the transcription and synthesis of a series of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β, etc.) in addition to the secretion of increased amounts of oxygen metabolites, chemokines, lysozyme, antimicrobial peptides, and proteases [35]. These cytokines act as “messengers” that not only exacerbate the local inflammatory response, but also actively recruit monocytes and neutrophils from the circulatory system to the lung lesions. These recruited immune cells effectively kill and eliminate invading pathogenic microorganisms through mechanisms such as the release of nitric oxide and the formation of neutrophil extracellular traps (NETs), reflecting the central defense role of lung macrophages in pathological processes such as ARDS [35, 36].
However, excessive inflammation can damage lung tissue, demonstrating the double-edged nature of the immune response.
To maintain immune balance, lung macrophages exhibit anti-inflammatory properties. Studies show that AMs suppress pro-inflammatory responses by phagocytosing dead cells during exposure to external stimuli, demonstrating anti-inflammatory effects [37]. Moreover, phagocytosis can in turn promote the secretion of anti-inflammatory factors by AMs, such as TGF-β, prostaglandin E2 (PGE2), and platelet-activating factor (PAF), further suppressing inflammatory responses [38]. The alveolar microenvironment is also actively involved in continuous signaling to promote immunosuppressive activity in AMs. AECs promote anti-inflammatory activity in AMs by producing IL-10 and TGF-β to activate integrin αvβ6 [37]. In addition, continuous CD200-mediated CD200 receptor signaling by type II AECs inhibits the c-Jun N-terminal kinase (c-JNK), p38 mitogen-activated protein kinase (p38), and extracellular signal-regulated kinase (ERK) signaling pathways in AMs, thereby suppressing the expression of proinflammatory cytokines [37]. These mechanisms precisely regulate the intensity and duration of inflammatory responses, aimed at maintaining the homeostasis of lung tissue, preventing unnecessary tissue damage, and thus playing a crucial regulatory role in the process of lung injury repair and regeneration.
Furthermore, lung macrophages dynamically switch between M1 and M2 phenotypes in response to environmental cues, representing two extremes of a functional spectrum following activation [13, 14]. However, persistent activation or dysregulation of M1 macrophages can lead to non-resolving inflammatory processes characterized by sustained production of pro-inflammatory mediators such as IL-1β, IL-6, and TNF-α. This prolonged inflammation contributes to chronic lung injury and impaired tissue healing, ultimately causing progressive tissue damage and dysfunction [39].
Conversely, M2 macrophages play a significant role in resolving inflammation and promoting tissue repair through the secretion of anti-inflammatory cytokines and growth factors such as IL-10 and TGF-β [40]. However, pathological or excessive activation of the M2 macrophage phenotype can lead to maladaptive tissue repair mechanisms, including lung fibrosis. Specifically, sustained or elevated production of TGF-β by M2 macrophages strongly promotes differentiation of fibroblasts into myofibroblasts, enhances excessive extracellular matrix deposition, and results in progressive pulmonary fibrosis. This aberrant fibrotic response significantly impairs lung function and contributes to chronic pulmonary disorders [40]. Thus, maintaining a delicate balance between M1 and M2 macrophage activation states is essential for proper resolution of inflammation and effective tissue repair.
Lung macrophages also play a significant role in tissue repair and regeneration. In the later stages of the inflammatory response, lung macrophages express increased amounts of IL-10 and arginase-1, which promote dissipation of inflammation and tissue repair [36, 41, 42]. As an anti-inflammatory cytokine, IL-10 could inhibit the production of pro-inflammatory cytokines and attenuate the inflammatory response, while arginase-1 promotes tissue repair by enhancing arginine metabolism – specifically, by converting L-arginine into ornithine and urea. Ornithine subsequently serves as a precursor for the synthesis of polyamines and proline. Proline, in particular, is a critical amino acid required for collagen production, which is essential for extracellular matrix formation and wound healing. In this way, the metabolic conversion of arginine facilitates the biosynthesis of key molecules necessary for tissue repair and regeneration [43]. By modulating macrophage polarization, lung macrophages are transformed into an anti-inflammatory and repair state. It also secretes growth and repair factors, such as TGF-β, VEGF (vascular endothelial growth factor) and PDGF, which stimulate the proliferation and migration of fibroblasts and endothelial cells, and promote angiogenesis and collagen deposition, thus accelerating the repair and regeneration process of lung tissues. It is very important for tissue repair and regeneration after lung injury [36].
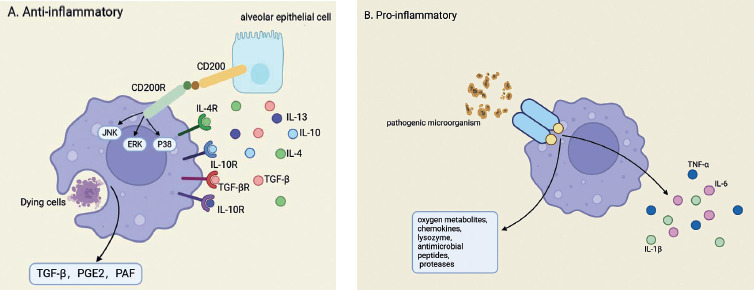
Moreover, amidst the onset and progression of lung injury, inflammatory and physicochemical insults evoke the activation of lung epithelial progenitor or stem cells. These cells promptly proliferate and differentiate, thereby replenishing the compromised cellular components and facilitating the regenerative processes within lung tissue. Central to this orchestrated response, lung macrophages assume a pivotal function by stimulating the Wnt signaling pathway, which serves as a pivotal catalyst for promoting lung repair and regeneration [44]. Figure 1 illustrates the anti-inflammatory and pro-inflammatory effects of lung macrophages.
Fig. 1
Anti-inflammatory and pro-inflammatory effects of lung macrophages. A) Anti-inflammatory response of alveolar macrophages (AMs). AMs suppress inflammation through several pathways. The binding of CD200 to CD200R inhibits the JNK, ERK, and p38 signaling pathways, reducing the production of pro-inflammatory cytokines. Meanwhile, IL-4, IL-10, IL-13 and TGF-β, secreted by AECs, activate IL-4R, IL-10R, IL-13R, and TGF-βR, further promoting anti-inflammatory activity. AMs also secrete TGF-β, PGE2, and PAF after engulfing dying cells, maintaining immune homeostasis. B) Pro-inflammatory and antimicrobial response of AMs. Upon encountering pathogenic microorganisms, AMs initiate a pro-inflammatory response. They release reactive oxygen metabolites, chemokines, lysozymes, antimicrobial peptides, and proteases to eliminate pathogens. This response also involves the production of key cytokines such as TNF-α, IL-6, and IL-1β, which recruit other immune cells to the site of infection. The coordinated action of these molecules is essential for microbial clearance but must be tightly regulated to prevent excessive inflammation
Antigen presentation and adaptive immunity
Alveolar macrophages, as dedicated antigen-presenting cells, possess the remarkable ability to sequester, process, and subsequently present antigens to T cells, thereby orchestrating a pivotal role in both innate and adaptive immune defenses of the host. In response to local lung tissue infections initiated by pathogen colonization, these macrophages harness PRRs on their surface to identify and phagocytose the invading pathogens. In cases of severity, they undergo further activation, presenting the internalized antigens to adaptive immune cells, effectively bridging the innate and adaptive immune responses. Of critical importance is the fact that the significant phenotypic plasticity allows AMs to maintain a dynamic balance between pro-inflammatory and anti-inflammatory responses. This phenotypic transformation triggers a cascade of immune reactions through the orchestrated release of signaling molecules, ultimately modulating both inflammatory injury and lung tissue repair processes. Moreover, lung macrophages are integral to the restoration of damaged inflammatory tissues, highlighting their dual role in both immune activation and subsequent resolution. They also collaborate seamlessly with other antigen-presenting cells, notably DCs, to amplify and sustain adaptive immune responses, ensuring a robust and coordinated defense against lung pathogens [45].
In adaptive immunity, the classical activation of M1 macrophages is crucial for initiating immune responses against pathogens. M1 macrophages release IL-1, IL-6, IL-12, IL-23, and TNF-α, facilitating pathogen clearance and T cell activation [46]. Conversely, M2 macrophages play a role in resolving inflammation and promoting tissue repair. M2 macrophages release IL-10 and TGF-β, modulating immune responses and promoting the transition from acute inflammation to tissue resolution and repair [45].
Role of lung macrophages in pulmonary diseases
Acute respiratory distress syndrome (ARDS)
Acute respiratory distress syndrome is a severe form of acute diffuse inflammatory lung injury that progresses rapidly and poses a significant threat to human life, with mortality rates in severe cases reaching 50-70% [35]. The hallmark features of ARDS include damage to AECs, an overwhelming inflammatory response, and increased permeability of the alveolar–capillary barrier. Diffuse alveolar damage (DAD) is frequently observed in autopsy studies [47].
ARDS can be triggered by a variety of insults, not only by infections but also by mechanical or chemical lung injuries, fat embolism, and other factors. In the acute phase, as key components of the innate immune system, AMs express PRRs to detect PAMPs and damage-associated molecular patterns (DAMPs). These cells play a pivotal role by releasing high levels of pro-inflammatory cytokines (such as IL-1β, IL-6, and TNF-α), which recruit neutrophils and other leukocytes to the lung, thereby exacerbating inflammation and contributing to further AEC damage [48-50]. Due to the excessive inflammatory response, the protective mechanisms fail to resolve the inflammation, leading to persistent tissue injury and chronic inflammation. During the recovery phase, AMs help modulate inflammation and promote tissue repair by clearing cellular debris, interacting with AECs, and secreting growth factors. However, in some patients, an overactive or dysregulated repair response may lead to pathological fibrosis. This fibrotic process can be partly attributed to the oxidative stress from high-flow oxygen therapy and an excessive M2 macrophage response that produces large amounts of TGF-β. TGF-β, in turn, activates fibroblasts and stimulates collagen deposition, ultimately resulting in lung fibrosis and long-term impairment of lung function [51]. Additionally, IMs contribute to the repair of the alveolar–capillary membrane by secreting angiogenic factors such as VEGF and trophic factors such as keratinocyte growth factor (KGF). While these factors support the reconstruction of the pulmonary microvascular network, they may also participate in pathological repair if the inflammatory process remains unchecked [51].
Thus, the failure of protective mechanisms in ARDS is twofold: persistent M1-driven inflammation leads to immune dysregulation, and an excessive M2-mediated repair response, driven by factors such as TGF-β, results in pathological fibrosis. Maintaining a balanced macrophage response is critical for effectively resolving inflammation without triggering deleterious fibrotic remodeling.
Asthma
Asthma is characterized by chronic airway inflammation, with clinical manifestations including immune cell infiltration, airway hyperresponsiveness, excessive mucus production, and airway remodeling, all of which contribute to restricted airflow and dyspnea. AMs play a crucial role in asthma pathogenesis by promoting allergic airway inflammation. They facilitate inflammatory cell recruitment and activation and secrete factors that induce structural cell thickening and airway remodeling [52]. Furthermore, AMs participate in the airway remodeling process by secreting factors such as IL-4, IL-13, profibrotic growth factors, TGF-β, and platelet-derived growth factor (PDGF), which promote the proliferation and migration of fibroblasts, as well as the synthesis and deposition of ECM [53, 54].
In addition to the role of AMs, eosinophils are recognized as a critical player in the inflammatory process of asthma. Eosinophils release cytotoxic granule proteins, cytokines, and lipid mediators that exacerbate airway inflammation, mucus hypersecretion, and tissue remodeling [55]. Importantly, the crosstalk between eosinophils and macrophages amplifies the inflammatory response; for instance, eosinophils produce IL-5, which can enhance macrophage activation, while activated macrophages secrete chemokines that further recruit eosinophils [56].
During acute asthma exacerbations, the number and activity of AMs increase significantly, thereby promoting inflammatory responses. Conversely, during remission, AMs contribute to the resolution of inflammation and tissue repair by suppressing excessive immune responses [52]. The dynamic polarization of AMs, shifting from a pro-inflammatory M1 state to an anti-inflammatory M2 state, is critical for asthma management and treatment strategy development. Modulating this polarization offers promising therapeutic avenues for controlling airway inflammation and improving outcomes in asthma patients.
Moreover, current research has identified distinct asthma phenotypes based on inflammatory cell profiles in induced sputum or BAL. For example, eosinophilic asthma – characterized by elevated eosinophils – is often responsive to targeted therapies such as anti-IL-5 agents (e.g., mepolizumab, benralizumab), anti-IL-4/IL-13 treatments (e.g., dupilumab), or anti-IgE therapy (e.g., omalizumab) [57]. Such phenotype-specific approaches underscore the importance of tailoring treatment strategies based on the dominant inflammatory pathways in individual patients.
Interstitial macrophages also play a significant role in the pathological process of asthma. In chronic asthma, interactions between IMs, airway smooth muscle cells (ASMCs), and fibroblasts contribute to persistent inflammation. IMs may aggravate inflammation by secreting pro-inflammatory factors such as TNF-α and IL-1β, yet they can also exert anti-inflammatory effects through alternative polarization [58]. ASMCs and fibroblasts are directly involved in ECM deposition and structural remodeling, leading to airway wall thickening and reduced elasticity. Furthermore, IMs indirectly influence airway remodeling by modulating ASMC phenotypes and promoting fibroblast activation into myofibroblasts, thereby contributing to airway fibrosis and the progression of structural changes [59].
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) is a complex, heterogeneous disorder characterized primarily by chronic airway inflammation, emphysema, and airflow limitation, rather than lung fibrosis being the main pathological feature [60, 61]. Although structural lung abnormalities, including some degree of fibrosis, can occur in COPD, fibrosis is not the predominant issue in most patients. Chronic inflammation is central to COPD pathogenesis, characterized by altered immune cell numbers and dysfunction. In COPD, AMs play a pivotal role in airway inflammation, particularly in smokers and patients with severe disease, where both AM number and activity are elevated. AMs process antigens – from pathogens to allergens – and present them to T cells, thereby initiating adaptive immune responses. However, in COPD, immune dysfunction often compromises AM function, impairing antigen clearance and promoting persistent inflammation and lung tissue damage [60, 62]. While AMs contribute to tissue repair, the therapeutic promotion of repair processes must be approached with caution in COPD. For example, agents targeting the TGF-β signaling pathway might enhance repair but could also exacerbate fibrotic remodeling if used indiscriminately [63].
Recent studies have demonstrated that COPD can be classified into distinct subtypes based on the cellular composition of induced sputum or BAL fluid. These subtypes include: 1) eosinophilic COPD – characterized by elevated eosinophils, often responsive to inhaled corticosteroids and anti-IL-5 therapies (e.g., mepolizumab, benralizumab) [64]; 2) neutrophilic COPD – characterized by high neutrophil counts, typically associated with a poor response to corticosteroids and potentially benefiting from alternative anti-inflammatory or antimicrobial strategies [65]; 3) mixed or paucigranulocytic COPD – where the inflammatory cell profile does not display a clear predominance, suggesting the need for personalized treatment approaches [65].
In addition to inflammation, AMs contribute to oxidative stress by releasing reactive oxygen species (ROS), which further amplify lung damage and impair repair mechanisms [66]. Furthermore, although IMs interact with fibroblasts to stimulate the production of profibrotic cytokines – such as TGF-β1 and PDGF – which drive fibroblast activation and differentiation into myofibroblasts, these fibrotic responses are generally secondary to the primary inflammatory processes in COPD [67].
Understanding these complex mechanisms – along with the identification of COPD subpopulations – underscores the necessity for personalized therapeutic strategies that effectively reduce inflammation while avoiding the risk of pathological tissue repair and fibrosis.
Influenza, COVID-19, and other respiratory infections
Alveolar macrophages play a crucial role in host defense against both viral and bacterial infections [68]. Particularly during influenza virus infection, infected alveolar type II cells and epithelial cells can release signals that attract macrophages to their location and promote phagocytosis [69]. Furthermore, in response to the activation of PRRs (TLRs), AMs are capable of producing a range of cytokines, including IL-6, IL-12, etc., which contribute to effectively limiting the spread of the virus [70]. This rapid immune response is essential for controlling viral infections and reducing immune-mediated damage to the host.
In the context of COVID-19, AMs also play a central role in the immune response. SARS-CoV-2 infection damages AECs, triggering AM activation. However, overactivation of AMs in severe COVID-19 cases can lead to a cytokine storm characterized by excessive production of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α [71]. This hyperinflammatory state contributes to acute lung injury and the progression to ARDS [72]. Additionally, neutrophils are heavily involved in severe COVID-19. They release NETs, which can further activate macrophages and amplify the inflammatory response, thereby exacerbating lung injury [36].
In addition to their role in infection control, AMs contribute to tissue repair and immune homeostasis. After the clearance of infections – including influenza and COVID-19 – AMs shift toward an M2 phenotype, secreting anti-inflammatory cytokines and growth factors, such as IL-10 and TGF-β, to promote tissue repair and regeneration [36]. IMs can also contribute to tissue repair and immune balance through the secretion of IL-10 and TGF-β. However, caution is warranted: an excessive M2 response may lead to overproduction of TGF-β, potentially driving pathological fibrotic remodeling [73]. In fact, post-COVID lung fibrosis, which affects approximately 8-10% of patients with severe SARS-CoV-2 infection requiring hospitalization, underscores the risk of promoting excessive tissue repair in this context. Early interventions in severe COVID-19 cases focus on reducing the pathological inflammatory reaction, particularly the cytokine storm, to prevent long-term complications such as fibrosis.
Moreover, AMs release ROS during infections to aid pathogen clearance. However, excessive ROS production can exacerbate tissue damage, highlighting the delicate balance between immune defense and tissue injury. Targeting macrophage activation pathways may enhance antiviral therapies by promoting viral clearance while minimizing immune-related tissue damage.
Concurrently, AMs maintain pulmonary homeostasis by phagocytosing dead or damaged cells. Their multifaceted role in regulating immune responses and facilitating tissue repair is essential for maintaining lung health.
Tuberculosis
Tuberculosis (TB), an ancient global pandemic dating back to prehistoric times, is caused by Mycobacterium tuberculosis (Mtb). The transmission of this disease typically occurs through the aerosolized droplet nuclei expelled by infected individuals during coughing [74, 75].
The early response of AMs to pathogens involves a complex array of signaling pathways aimed at controlling microbial growth while simultaneously triggering inflammatory responses to recruit other immune cells to the site of infection. In the context of TB, the interaction of AMs with Mtb is particularly critical. These macrophages are among the first cells to encounter the bacteria, and their initial response is pivotal in shaping the subsequent adaptive immune response necessary for controlling the infection [75]. After phagocytosis, AMs and DCs present TB antigens to immune cells. Activated effector CD4+ Th1 cells release IFN-γ and TNF-α, which enhance AMs bactericidal activity. This response is vital for controlling Mtb infection and resolving inflammation [76].
Despite the immune defense initiated by AMs, Mtb employs several strategies to evade macrophage-mediated immunity and persist within host cells. Mtb can inhibit phagolysosome formation and acidification, rendering AMs less effective in killing the bacteria. Additionally, it can tolerate the toxic effects of ROS and suppress essential processes such as autophagy and apoptosis, ensuring survival within macrophages [77, 78].
A critical component of the host defense against TB is the formation of granulomas – organized aggregates of macrophages, lymphocytes, and other immune cells that contain Mtb and prevent its dissemination [79]. TNF-α plays a key role in maintaining granuloma integrity. However, biological therapies targeting TNF-α, such as infliximab, adalimumab, and etanercept used in rheumatoid diseases, can disrupt granuloma structure and increase susceptibility to TB and other mycobacterial infections [80]. Therefore, immunomodulatory treatments in patients at risk must be used with caution.
During pulmonary TB infection, IMs help suppress excessive inflammatory responses by secreting IL-10, an anti-inflammatory cytokine that downregulates the production of pro-inflammatory cytokines, thereby reducing tissue damage and facilitating lesion repair [81]. This regulatory function is crucial for preventing tissue damage caused by TB and facilitating the repair of lesions. Recent studies have also highlighted the importance of the transcription factor NRF2 in the early response of AMs to Mtb. Infected AMs upregulate an NRF2-driven antioxidant transcriptional response that is distinct from the pro-inflammatory responses typically associated with macrophage activation [75]. Moreover, the early response of AMs to Mtb appears to be less inflammatory than previously thought, which may have implications for the overall host response. The NRF2-mediated response in AMs could potentially hinder the initiation of a robust adaptive immune response, critical for long-term TB control [75]. Given the complexity of Mtb – macrophage interactions, therapeutic strategies targeting AMs offer promising avenues for TB management. Modulating AM polarization from an NRF2-dominant antioxidant state to a more pro-inflammatory, bactericidal state could enhance Mtb clearance. In addition, strategies aimed at restoring phagolysosome function and promoting autophagy through host-directed therapies (HDT) – such as the careful use of corticosteroids or agents that modulate the NF-κB pathway – may improve TB treatment efficacy [82]. However, caution is warranted with immunomodulatory drugs, particularly TNF-α inhibitors, as they may adversely impact granuloma integrity and TB clinical course [83].
Lung cancer
Lung cancer is a leading cause of cancer-related mortality worldwide, accounting for only 18% of all cancer diagnoses but possessing one of the lowest 5-year survival rates among all malignant neoplasms. Lung macrophages play a complex role in lung cancer, often polarized by the tumor microenvironment to become tumor-associated macrophages (TAMs), which exert a crucial influence on the development and progression of lung cancer [84].
Research indicates that in the early stages of lung cancer, M1-type macrophages predominantly exist, but as the tumor progresses, there is a phenotypic shift from M1 to M2, with TAMs in the middle and late stages typically exhibiting M2 polarization characteristics, which are associated with tumor promotion and immune suppression functions [85]. M2 macrophages, through the secretion of IL-10 and other immunosuppressive molecules, can activate tumor stem cells and inhibit the activity of T cells, thereby reducing the immune system’s ability to attack tumors and promoting tumor progression [86, 87]. Furthermore, M2 macrophages also aid in the invasion of tumor cells into surrounding tissues and the basement membrane by producing matrix metalloproteinases (MMPs) and other proteases, promoting the metastasis and invasion of lung cancer. They are also involved in regulating tumor angiogenesis, promoting the formation of new blood vessels through the secretion of factors such as VEGF, providing oxygen and nutrients for the tumor, thus supporting tumor growth and survival [84].
Interstitial macrophages within the tumor microenvironment can also interact with cancer cells to secrete MMPs, particularly MMP-2 and MMP-9, which facilitate the degradation of the ECM, thereby supporting the invasion and metastasis of cancer cells [88]. TAMs promote angiogenesis by regulating MMPs, breaking down the basement membrane, and secreting pro-angiogenic factors, cytokines, and chemokines, which in turn promote the formation of the tumor vascular network and enhance the tumor’s resistance to therapy [89].
In recent years, therapeutic strategies targeting TAMs have become a focal point in lung cancer research. For instance, the use of small molecule drugs or antibodies to block the recruitment of TAMs or to reprogram them into M1-type macrophages enhances their anti-tumor capabilities. One approach involves targeting the CD47-SIRPα (signal-regulatory protein a) axis, which can prevent the expression of CD47 by healthy and cancerous cells. CD47, when bound to SIRPα on macrophages, inhibits phagocytosis, thereby suppressing the anti-tumor activity of macrophages [90]. Furthermore, activating TAMs alongside T cells through combined immunotherapy strategies is an important direction in current research, aiming to achieve a more effective anti-tumor immune response [91].
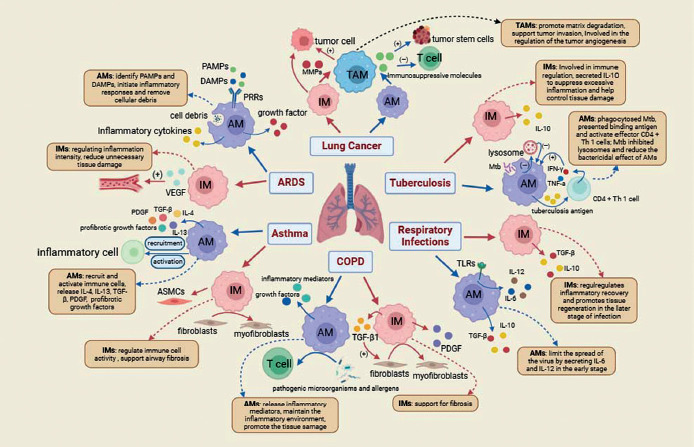
In summary, the role of TAMs in lung cancer is multifaceted; they are not only involved in tumor promotion and immune evasion but also represent potential therapeutic targets. Gaining an in-depth understanding of the specific mechanisms by which TAMs function in lung cancer is of significant importance for the development of novel treatment strategies. The Figure 2 summarizes the role of AMs and IMs in pulmonary diseases.
Fig. 2
The roles of AMs and IMs in pulmonary diseases. This figure illustrates the distinct roles of AMs and IMs in various pulmonary diseases, including ARDS, asthma, COPD, lung cancer, TB, and other respiratory infections. AMs and IMs exhibit remarkable functional plasticity and coordinate immune responses, tissue repair, and immune regulation. In ARDS, AMs initiate acute inflammation through recognition of PAMPs and DAMPs, while IMs promote recovery by regulating inflammation and supporting angiogenesis. In asthma, AMs recruit immune cells through IL-4 and IL-13 secretion, while IMs interact with fibroblasts to promote airway remodeling. In COPD, AMs maintain chronic inflammation, and IMs contribute to fibrosis by interacting with structural cells. In lung cancer, TAMs, which originate from both AMs and IMs, promote tumor progression, immune suppression, and metastasis by producing MMPs and VEGF. During TB infection, AMs combat Mtb through phagocytosis and antigen presentation, while IMs modulate excessive inflammation through IL-10 secretion. In respiratory infections, AMs control viral spread through IL-6 and IL-12 release, and IMs promote tissue repair through IL-10 and TGF-β. This figure highlights the complementary roles of AMs and IMs in maintaining pulmonary homeostasis and underscores their significance as potential therapeutic targets in pulmonary diseases
The impact of trained immunity on the function of lung macrophages
Trained immunity, as a form of innate immune memory, holds significant relevance in the context of lung macrophage research. This phenomenon refers to the enhanced and accelerated response that trained immune cells can mount against homologous or even heterologous pathogens upon secondary encounters, even in the absence of adaptive immune components such as T and B cells [92].
In the alveoli, macrophages serve as the first line of defense, constantly exposed to inhaled particles and microorganisms. This exposure imprints a form of memory on the macrophages, known as trained immunity, which enhances their response to subsequent challenges. The mechanisms of trained immunity involve epigenetic reprogramming and metabolic reprogramming, induced by various stimuli, including microbial components and certain immunomodulatory agents [93].
Epigenetic reprogramming is a pivotal mechanism in the process of trained immunity [94]. During this process, macrophages undergo alterations in chromatin structure, encompassing modifications in histone modifications and DNA methylation status. These changes facilitate the opening of chromatin, thereby promoting rapid expression of pro-inflammatory factors and the formation of memory. In the context of trained immunity, it has been observed that well-trained lung macrophages, following an initial stimulus, exhibit certain epigenetic modifications that enable faster expression of related effector genes upon re-stimulation. Notably, these modifications include non-permanent histone modifications associated with gene activation, such as H3K4 monomethylation and trimethylation, as well as H3K27 acetylation [95]. These epigenetic marks, detected through various methodologies such as chromatin immunoprecipitation (ChIP) and the assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq), lead to chromatin opening at the promoters of genes encoding pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF. This chromatin accessibility is correlated with protection against subsequent infections [96].
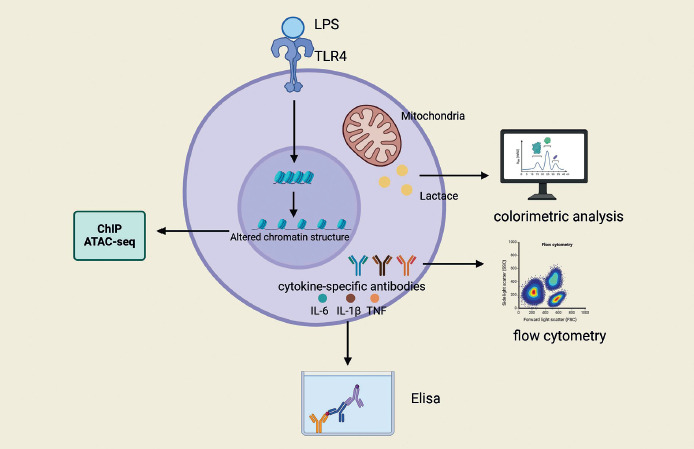
Metabolic reprogramming emerges as another cornerstone in the establishment and maintenance of the trained immunity state. Studies have demonstrated that exposure to lipopolysaccharide (LPS) can induce trained immunity in lung macrophages, leading to significant alterations in the composition of metabolites and lipids, including a marked increase in the levels of certain ceramides, phosphatidyl ethanolamines, sphingomyelins, and phosphatidylcholines, while the levels of triglycerides are significantly reduced [93]. These changes potentially have a direct impact on membrane receptor signaling, thereby modulating the functional state of lung macrophages and enhancing their activity following pneumococcal invasion. Concurrently, lung macrophages in a state of trained immunity exhibit reduced basal metabolic activity, as evidenced by decreased oxygen consumption rates (OCR) and extracellular acidification rates (ECAR), and this metabolic signature remains significantly lower during a secondary bacterial attack [93]. Furthermore, the induction and sustenance of trained immunity involve multiple metabolic pathways, including glycolysis, oxidative phosphorylation, the tricarboxylic acid (TCA) cycle, and the metabolism of amino acids and lipids [97]. These metabolic pathways not only provide energy and biosynthetic precursors but also directly regulate epigenetic mechanisms by producing intermediate metabolites that serve as substrates or cofactors, thereby facilitating the expression of inflammatory genes. Figure 3 summarizes the research methods in training immunity.
Fig. 3
Research methods for training and immunity. Training immunity can be assessed by different methods. Training immunity can be assessed by different methods. Altered chromatin structure can be detected with the help of ChIP, ATAC-seq, etc. Flow cytometry can be used to quantify the production of proinflammatory cytokines in LPS innate immune cells, such as TNF and IL-6. ELISA can be used to measure proinflammatory cytokine responses (IL-6, TNF, and IL-1β) both in vivo and in vitro. The trained cells have increased mitochondrial activity in vitro and can be assessed by colorimetric assays
The effects of training immunity on lung macrophage function are being actively explored, especially in the context of diseases such as ARDS, asthma and COPD. Current research indicates that AMs can be “trained” by viral infections to enhance their phagocytic activity and cytokine production, which aids in the clearance of pathogens and contributes to the resolution of inflammation [46, 98, 99]. This trained state can persist for an extended period, providing long-term protection against a variety of pulmonary diseases.
The concept of trained immunity extends beyond infectious diseases and shows promise in the context of cancer immunotherapy [46]. Studies have found that β-glucan-induced trained immunity has antitumor effects. This antitumor effect of trained immunity is associated with the transcriptional and epigenetic reprogramming of granulopoiesis, and it is also related to the reprogramming of neutrophils toward an antitumor phenotype; this process requires type I IFN signaling and is independent of the host’s adaptive immunity [100].
However, trained immunity can also lead to adverse effects under certain circumstances. Researchers have transferred lung macrophages, which were isolated five days after in vivo training with LPS, into initial recipients via intratracheal administration. An increase in bacterial load and pulmonary inflammation was observed in the recipients 24 hours later, indicating that trained lung macrophages may lead to increased pulmonary inflammation and impaired bacterial clearance in specific environmental and disease contexts [93]. Furthermore, under conditions of chronic inflammation, trained immunity may be maladaptive, contributing to the progression of excessive inflammation and autoinflammatory syndromes, and aiding in disease development [101].
In summary, the impact of trained immunity on the function of lung macrophages is complex and multifaceted. It has the potential to enhance resistance to infections but may also lead to adverse reactions under certain conditions. This duality has significant implications for the development of novel therapeutic strategies for respiratory diseases. Therefore, the study of trained immunity is a complex endeavor that requires a deep investigation into its metabolic pathways and a consideration of its mechanisms and impacts within the context of various disease states. This is essential not only to comprehend the intricacies of trained immunity but also to enhance pulmonary immune defenses and to harness its potential in clinical applications, thereby providing novel strategies for clinical intervention.
Vaccinations significantly impact trained immunity by inducing long-term functional reprogramming of innate immune cells, including macrophages [102]. For example, the Bacillus Calmette-Guérin (BCG) vaccine induces trained immunity by epigenetic and metabolic reprogramming of macrophages, enhancing their capacity to respond rapidly and robustly to subsequent unrelated pathogens (heterologous protection). Similar mechanisms are observed following influenza and pneumococcal vaccinations, highlighting potential clinical strategies for harnessing macrophage-trained immunity in protecting against diverse pulmonary infections [103].
Therapeutic targeting of lung macrophages
Gene therapy and precision medicine
Emerging therapeutic strategies based on gene editing and molecular targeting of lung macrophages are becoming potent tools for the treatment of lung diseases. Research has demonstrated that through gene-editing technologies such as CRISPR (clustered regularly interspaced short palindromic repeats)–Cas9 (CRISPR associated protein 9), the deletion or insertion of specific genes can reprogram macrophages from an immunosuppressive state (e.g. M2 phenotype) to an immunoactivated state (e.g. M1 phenotype), thereby enhancing their antitumor capabilities [104]. By precisely modifying gene expression within macrophages, scientists can study the impact of specific genes on macrophage function, altering cellular behavior and enhancing their ability to clear specific pathogens or modulate immune responses in disease contexts [105].
Molecular targeted therapies intervene in specific molecular pathways of lung macrophages to modulate their functions. These treatments utilize small molecule drugs or monoclonal antibodies that target molecular nodes such as cytokine receptors, enzymes, or transcription factors, influencing macrophage activation, polarization, and effector functions. Studies have shown that targeting the colony-stimulating factor-1 receptor (CSF-1R) can regulate the survival and differentiation of macrophages, which may have potential therapeutic benefits for certain lung diseases, including pulmonary fibrosis or specific types of pneumonia [106].
Possible limitations or challenges in clinical application
Targeted therapies based on lung macrophages play a pivotal role in disease treatment; however, their clinical application may still face certain limitations and challenges. In targeted therapy, if the drug delivery system lacks accuracy, drugs may be distributed to off-target cells, leading to unintended side effects. To address this issue, researchers have developed a strategy using folic acid-functionalized exosomes (FA-Exo) to achieve targeted delivery to M1-type macrophages, thereby increasing drug accumulation at the site of pulmonary disease and minimizing off-target effects [107]. The design of drug delivery systems must take into account not only the distribution and metabolism of the drug within the body but also the precision with which the drug is delivered to the target cells. In this regard, a team has successfully encapsulated mRNA encoding a single-chain antibody against IL-11 within optimized lipid nanoparticles (LNPs). This approach enhances targeted delivery to specific tissues or cells and minimizes adverse reactions [108]. However, even if drugs are precisely delivered to the target cells, they may still affect off-target cells due to the drug’s inherent side effects. Thus, ensuring the specificity of targeted drugs remains one of the challenges currently faced in the field [109]. Although researchers have made some progress in addressing the challenges of targeted therapy, several issues still require ongoing attention to resolve. These include ethical considerations related to targeted therapy, the development of drug resistance, long-term effects and safety, and the cost and accessibility challenges caused by the complex drug design and delivery systems. By addressing these concerns, we can maximize the therapeutic potential of these treatments and improve patient outcomes.
Challenges and future directions
Unanswered questions in macrophage biology
Human alveolar macrophages (HAMs) are a rare and elusive cell population, residing deep within the lung and posing significant challenges and risks for direct acquisition from the human body. The limited availability of HAMs, coupled with their propensity to undergo phenotypic changes and lose their original functions and characteristics during in vitro culture, restricts their application in laboratory research. Although it is widely acknowledged that lung macrophages exhibit heterogeneity, this diversity complicates their classification and study. The extent to which heterogeneity reflects different origins, micro-anatomical specialization, or historical remnants of various injuries to the epigenetic and phenotypic landscape remains unclear [8]. Studies have indicated that both embryonic and adult precursors give rise to macrophages with nearly identical transcriptional profiles at birth when they occupy empty niches, yet these cells display a high degree of heterogeneity and dynamic changes in the context of aging, injury, and disease in the lung [8]. More sophisticated lineage tracing methods are required to further understand their developmental origins and lineages.
Technological and methodological challenges
Despite the new perspectives offered by technologies such as single-cell sequencing for studying lung macrophages, current research has predominantly focused on the level of gene expression, with less attention given to protein function and regulation. The complexity and cost of these technologies also limit their widespread application, necessitating the development of new techniques and methods to overcome these limitations [110]. Additionally, accurately detecting and assessing the function and status of lung macrophages in vitro, as well as translating research findings into clinical applications, remain pressing issues that require close collaboration and joint efforts between researchers and clinicians.
Potential for clinical translation
Macrophages play a multifaceted role in respiratory diseases, offering opportunities for the development of novel therapeutic strategies. In addition to modulating the polarization states of macrophages to improve disease outcomes through the regulation of inflammatory responses and tissue repair processes, macrophage-targeted therapies may also exert their effects by reducing pathological inflammation and promoting the repair of lung tissue [111]. Despite the considerable potential, translating these research findings into clinical applications still faces challenges, including ensuring the safety, efficacy, and accessibility of drugs.
To achieve the clinical translation of macrophage-targeted therapies, a deeper understanding of the specific mechanisms of macrophages in different disease states is required. Furthermore, the development and validation of new therapeutic tools that can precisely modulate macrophage functions are necessary. The design and execution of clinical trials must also take into account individual differences and disease heterogeneity to ensure personalized and optimized treatment strategies.
Conclusions
Lung macrophages are essential components of the pulmonary immune system, demonstrating irreplaceable importance in maintaining lung homeostasis, defending against pathogen invasion, promoting tissue repair, and finely regulating immune responses. They efficiently clear pathogens and foreign substances, such as bacteria and viruses, modulate inflammatory responses, present antigens, and actively participate in tissue repair and regeneration processes, thereby effectively protecting the lungs from damage. Furthermore, lung macrophages play a key role in the onset and progression of pulmonary diseases such as ARDS, asthma, COPD, and influenza, with their functional abnormalities often closely associated with disease progression. With the rapid development and application of emerging therapeutic approaches such as gene editing and molecular targeting, our understanding of the heterogeneity, developmental origins, and functional states of lung macrophages has reached unprecedented depths.
Research on lung macrophages holds significant potential for the treatment of pulmonary diseases. Understanding the molecular mechanisms and pathological functions of AMs can lead to the identification of new targets and strategies for disease prevention, early diagnosis, and precision therapy. This research is expected to improve treatment outcomes and quality of life for patients with pulmonary diseases and provide important safeguards for lung health.
Recent years have witnessed the development of novel techniques and model systems, which have helped to overcome previous limitations in lung macrophage research. Real-time intravital imaging technology has allowed researchers to visualize the motility of AMs within and between alveoli and better understand their role in maintaining immune homeostasis [112]. This breakthrough has shed light on the dynamic behavior of AMs in live tissues, addressing previous knowledge gaps. Furthermore, the advent of a dual homologous recombination genetic fate-mapping technique (Dre-rox and Cre-loxP systems) has enabled the construction of specialized tool mice, providing deeper insights into the specific functions of pleural cavity macrophages [113]. This technology offers new therapeutic opportunities for addressing organ inflammation and promoting tissue repair. In addition, the development of a new cell culture model of HAMs has been a major milestone. Researchers have successfully created culture conditions using pulmonary lipids and lung-related cytokines to transform blood-derived monocytes into alveolar macrophage-like (AML) cells in vitro [114]. This model serves as an invaluable tool for the study of pulmonary inflammatory diseases and may lay the groundwork for the discovery of novel therapeutic approaches. The integration of these advanced technologies and model systems has not only enhanced our understanding of the complex roles of lung macrophages in health and disease but also opened new avenues for therapeutic intervention in pulmonary disorders.
Despite the significant progress made, the field of lung macrophage research still faces many challenges. Future research will need to employ interdisciplinary comprehensive methods and technologies to more precisely elucidate the molecular mechanisms and pathological functions of AMs. In-depth exploration of their origins, polarization processes, functional regulatory mechanisms, and complex interaction networks with other pulmonary immune cells is essential. This effort is expected to provide new targets and strategies for disease prevention, early diagnosis, and precision therapy, thereby improving treatment outcomes and quality of life for patients with pulmonary diseases and contributing significantly to the protection of lung health.