Introduction
Sepsis is a fatal inflammatory disease caused by infection, and frequently leads to multi-organ dysfunction. Acute kidney injury (AKI) is a significant comorbidity in sepsis, and sepsis is one of the major causes of AKI [1-3]. Statistically, 25-75% of AKIs worldwide are associated with sepsis or infectious shock [4]. AKI is a renal disorder characterized by a sudden reduction in kidney function, often presenting with oliguria or anuria, and elevated lactate, serum creatinine (SCr), and blood urea nitrogen (BUN) levels. The mortality rate can reach up to 74.5% [5]. The mortality rate of patients with sepsis combined with AKI is much higher than that of patients with sepsis alone [6]. The diagnostic marker SCr suffers from limitations. Its relatively long half-life and the existence of renal functional reserve mean that SCr elevation only occurs after a significant (25-50%) loss of renal function, thus hindering timely assessment of the glomerular filtration rate [7]. Furthermore, SCr primarily responds to glomerular function, whereas the site of injury in most AKIs is initially in the renal tubules. Therefore, the clinical diagnosis of sepsis-AKI lacks markers with higher accuracy and specificity.
Numerous long-chain non-coding RNAs (lncRNAs) and microRNAs (miRNAs) have been extensively studied in sepsis-induced AKI, and they have been shown to play critical roles [1, 8]. For example, lncRNA RMRP promotes sepsis-induced AKI by up-regulating DDX5 through miR-206 [9]. Up-regulation of miR-16-5p expression can significantly aggravate renal injury and apoptosis in sepsis-associated AKI [10]. miR-874-3p is identified as a potential biomarker for AKI and inhibits disease development via targeting MSRB3 [11]. Inhibition of SNHG14 suppresses NF-κB and the production of inflammatory cytokines via miR-373-3p [12]. LncRNA FENDRR (fetal lethal non-coding developmental regulator RNA), located at 16q24.1, serves as a marker for many malignancies [13]. While studies on FENDRR cover a range of diseases and pathologies, there is a lack of research specifically focused on FENDRR in the context of AKI or sepsis. During the literature search, we noted that FENDRR was significantly associated with AKI due to extracorporeal shock wave lithotripsy and was a biomarker for ESWL-AKI [14]. Moreover, the functions of FENDRR are closely intertwined with the biological processes underlying sepsis, positioning it as a potential regulatory factor in sepsis-related mechanisms [15]. Therefore, we hypothesized that FENDRR might be involved in sepsis-induced AKI by affecting cellular inflammation. Lnc-RNAs play a key role as competitive RNAs for miRNAs in many diseases [16]. It is plausible that FENDRR may engage in multiple facets of the inflammatory response by targeting specific miRNAs [17]. Studies have suggested that overexpression of miR-3614-5p attenuates LPS-induced inflammatory injury [18]. The expression of miR-3614-5p is closely associated with the cellular inflammatory response, suggesting that FENDRR may play a role as a competitive RNA for miR-3614-5p in sepsis-induced AKI.
In summary, FENDRR is differentially expressed in sepsis and AKI, and FENDRR/miR-3614-5p appears to play a role in cellular inflammation. However, whether lncRNA FENDRR can directly bind to miR-3614-5p and jointly participate in sepsis-induced AKI still needs to be verified. This study investigated the potential value of lncRNA FENDRR as a diagnostic marker for AKI. It aimed to provide a reference for finding new therapeutic targets for AKI.
Material and methods
Ethical statement
The present study was conducted following the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. Subjects signed an informed consent form before enrollment. The study protocol was approved by the Ethics Committee (approval No. 2020021) and strictly followed the principles of the Declaration of Helsinki.
Study subjects
One hundred and seventy-five patients with sepsis- induced AKI treated at Beijing Rehabilitation Hospital, Capital Medical School from August 2021 to December 2023 were included. All patients were admitted to the intensive care unit (ICU). Eighty-five patients with AKI triggered by sepsis served as the AKI group (patients were assessed daily for AKI after admission, and those who developed AKI within 7 days of enrollment were included). The remaining patients who did not trigger AKI totaled 90 patients and served as the control group. All patients with sepsis met the third international consensus definition of sepsis and septic shock as revised in 2016 [19]. The diagnosis of AKI secondary to sepsis was confirmed according to the Kidney Disease Improving Global Outcomes (KDIGO) 2012 criteria [20]. Sepsis-induced AKI patients were screened based on SCr levels (an increase of > 26.5 µmol/l within 48 hours or an increase to ≥ 1.5 times the baseline value within 7 days). The CONSORT flow diagram is shown in the Supplementary Figure 1.
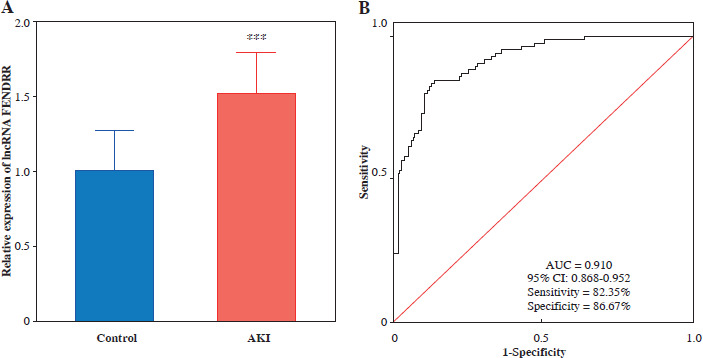
Fig. 1
FENDRR significantly increased in acute kidney injury (AKI). A) The expression of FENDRR was up-regulated in the serum of AKI patients (n = 85) compared to the control group (n = 90). B) Receiver operating characteristic (ROC) curves were analyzed for the diagnostic value of lncRNA FENDRR in AKI. ***p < 0.001 vs. controls
Exclusion criteria: 1) patients diagnosed with acquired immune deficiency syndrome; 2) patients with malignancies; 3) patients suffering from renal diseases, including those with end-stage renal disease maintained on hemodialysis and those with a history of renal transplantation; 4) patients who have been exposed to nephrotoxic medications, including those who received intravenous treatment with β-lactam antibiotics (cefotaxime, piperacillin sodium/tazobactam, cefazolin, and ceftazidime), aminoglycoside antibiotics (amikacin and gentamicin), immunosuppressants (cyclosporine), vancomycin, and acyclovir within 5 days; 5) AKI caused by definite non-septic factors; 6) pregnant women or lactating women; and 7) patients with incomplete clinical data.
Clinical sample collection and clinical indicators
The initial blood sample collected after diagnosis of sepsis was used for experimental index analysis. Venous blood samples (5 ml) were collected from all patients into EDTA anticoagulant tubes. The blood was centrifuged at 4°C, 3000 g for 15 min to collect the supernatant, which was then stored at –80°C. Procalcitonin (PCT), SCr, and BUN were measured using kits (Keygenbio, Jiangsu, China).
Cell model construction and cell transfection
The human renal proximal tubule cell line HK-2 (Cell Bank, Chinese Academy of Sciences, Shanghai, China) was used. MEM complete medium (Procell, Wuhan, China) containing 10% fetal bovine serum, 100 µg/ml streptomycin, and 100 U/ml penicillin was used. The medium was placed in an incubator at 37°C with 5% CO2. The culture medium was changed every 2 days, and the cells were passaged at a 1 : 2 ratio approximately every 3 days. When the cell density reached 70% to 90%, cells were digested using 0.25% trypsin (Procell, Wuhan, China) without EDTA, and the resulting cell suspension was transferred to a new culture dish for further culture or cryopreservation. To construct a sepsis-induced AKI cell model, HK-2 cells were treated with 10 µg/ml lipopolysaccharide (LPS, Solarbio, Beijing, China) for 6 hours [21]. HK-2 cells not induced by LPS were used as a control. Empty vectors were used with pcDNA3.1 (Sangon, Shanghai, China). Transfection vectors included small interfering RNA for lncRNA FENDRR (si-FENDRR) and negative control (si-NC). The plasmids were transfected into HK-2 cells using Lipofectamine 2000 (Invitrogen, CA, Carlsbad, USA) transfection reagent.
Dual luciferase report
Binding sites for lncRNA FENDRR and miR-3614-5p were predicted using the online database ENCORI. Recombinant plasmids were obtained by ligating wild-type (WT) and mutant (MUT) lncRNA FENDRR sequences containing the miR-3614-5p binding site into the pmir-GLO dual luciferase reporter vector (Promega, Madison, WI, USA). The miR mimic (to increase miR-3614-5p levels in cells), mimic NC (negative control for the miR mimic), miR inhibitor (inhibits the activity of miR-3614-5p), and inhibitor NC (negative control for the miR inhibitor) were transfected with the plasmids into HK-2 cells. The study was structured across eight experimental groups: 1) WT + mimic NC; 2) WT + miR mimic; 3) WT + inhibitor NC; 4) WT + miR inhibitor; 5) MUT + mimic NC; 6) MUT + miR mimic; 7) MUT + inhibitor NC; 8) MUT + miR inhibitor. The Renilla luciferase assay (Promega) was performed according to the instructions, and the relative fluorescence activity was calculated.
Cell viability and apoptosis assay
HK-2 cells in the logarithmic growth phase were collected and counted after digestion with trypsin to prepare a cell suspension at a concentration of 5 × 104 cells/ml. A cell suspension (100 µl) with 3 replicates per group was added to each well of 96-well plates. After 0, 24, 48, and 72 h of incubation, 10 µl of Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) solution was added to each well and incubated for 2 hours away from light. The 450 nm absorbance values were determined using a microplate reader (Tecan, Mannedorf, Switzerland).
Apoptosis was determined using an Annexin V-FITC/PI kit (Elabscience, Wuhan, China). The blank control and positive control were prepared using the binding buffer and cell apoptosis positive control kit (Beyotime, Shanghai, China), respectively. Transfected cells were collected, digested with trypsin, and resuspended in binding buffer to achieve a concentration of 1 × 105 cells/ml. Annexin V-FITC and PI reagent were added to each sample according to the instructions, and the samples were incubated in the dark for 15 minutes. The apoptotic capacity was analyzed using flow cytometry (FACSCalibur, BD, USA).
Determination of cellular MDA and ROS content
Malondialdehyde (MDA) levels were detected using the Lipid Peroxidation MDA Assay Kit (Beyotime). Reactive oxygen species (ROS) were detected using the Reactive Oxygen Specific Assay Kit (Beyotime).
Enzyme-linked immunosorbent assay (ELISA)
Inflammatory cytokines tumor necrosis factor α (TNF-α), interleukin (IL)-1β, IL-6, NGAL, and KIM-1 were detected using an ELISA kit (mlbio, Shanghai, China).
Real-time qPCR
Total RNA was extracted using TRIzol (Invitrogen). RNA with an OD260/OD280 result close to 2.0 was used for reverse transcription into cDNA with a Reverse Transcription Kit (Takara, Kusatsu, Japan). A 20 µl qPCR reaction was prepared according to the instructions of the SYBR Premix Ex Taq II kit (Takara). The qPCR program was 95°C, 30 s; (95°C, 5 s; 60°C, 30 s) × 40 cycles; 60°C, 1 min; 4°C hold. The relative expression levels of lncRNA FENDRR (F: GACAGCCTTCGGATCTAGGC, R: GTTGGCTTTCTGGTGCTGTG) and miR-3614-5p (F: GTTCTGTCTTGGGCCACTTG, R: ACACCAAGATCTGAAGGCTAC) were sequentially detected using a fluorescence quantitative PCR instrument (Applied Biosystems, Foster City, California, USA). The 2ΔΔCt method was performed using GAPDH (F: GTCTCCTCTGACTTCAACAGCG, R: ACCACCCTGTTGCTGTAGCCAA) and U6 (F: CTCGCTTCGGCAGCACAT, R: TTTGCGTGTCATCCTTGCG) as an internal reference.
Data analysis
SPSS 23.0 was used to analyze the experimental data. The independent t-test or two-way ANOVA was used to compare the significance of data between groups. Count data were expressed using examples. A ROC curve was used to evaluate the diagnostic value of lncRNA FENDRR in AKI. Logistic analysis was used to analyze the influencing factors for the occurrence of AKI in sepsis patients. Numerical transformations in logistic analysis were divided by the mean. P < 0.050 was considered statistically significant.
Results
Comparison of baseline data of enrolled subjects
The control group consisted of 44 males and 46 females with a mean age of 47.44 ±13.50 years. The AKI group consisted of 36 males and 49 females with a mean age of 45.79 ±16.36 years. There was no significant difference in age, gender, body mass index (BMI), heart rate, MAP, or source of infection between the control group and the AKI patient group (p > 0.050). PCT, BUN, SCr, APACHE II, SOFA, lactate, and length of hospital stay were significantly higher in the AKI group than in the control group (p < 0.050, Table 1). The comparison of different degrees of renal injury with indicators is shown in the Supplementary Table 1.
Table 1
Comparison of clinical data between septic acute kidney injury (AKI) and controls
Clinical value of serum FENDRR in AKI
Significantly higher expression levels of FENDRR were observed in AKI patients compared with controls (p < 0.001, Fig. 1A). The diagnostic significance of serum FENDRR in AKI was evaluated using the ROC curve. The results showed that FENDRR had significant diagnostic value in differentiating between control and AKI patients, with an AUC of 0.910 (95% CI: 0.868-0.952). The sensitivity and specificity were 0.824 and 0.867, respectively (Fig. 1B).
Binary logistic regression analysis was performed for PCT, BUN, SCr, APACHE II, SOFA, lactate, length of hospital stay, and FENDRR. Logistic regression was used to calculate the odds ratios (ORs) to assess risk factors, with OR > 1 or OR < 1 indicating whether the indicators act as risk or protective factors, respectively. The results showed that PCT (OR = 2.781, 95% CI: 1.223-6.324, p = 0.015), BUN (OR = 2.349, 95% CI: 1.059-5.209, p = 0.036), SCr (OR = 3.156, 95% CI: 1.370-7.272, p = 0.007), SOFA (OR = 3.440, 95% CI: 1.545-7.659, p = 0.002), lactate (OR = 2.974, 95% CI: 1.339-6.490, p = 0.007), SOFA (OR = 3.440, 95% CI: 1.545-7.659, p = 0.002) and FENDRR (OR = 9.216, 95% CI: 4.001-21.231, p < 0.001) are risk factors for AKI (Table 2). PCT (r = 0.627, p < 0.001), BUN (r = 0.706, p < 0.001), SCr (r = 0.838, p < 0.001), APACHE II (r = 0.747, p < 0.001), SOFA (r = 0.856, p < 0.001), lactate (r = 0.671, p < 0.001) and length of hospital stay (r = 0.331, p = 0.002) were positively correlated with FENDRR in AKI patients (Table 3).
Table 2
Logistic analysis of independent factors influencing the occurrence of acute kidney injury (AKI) in patients with sepsis
Table 3
Correlation analysis between lncRNA FENDRR and clinical indicators in acute kidney injury (AKI) patients
Silencing FENDRR attenuates cellular inflammatory damage and oxidative stress
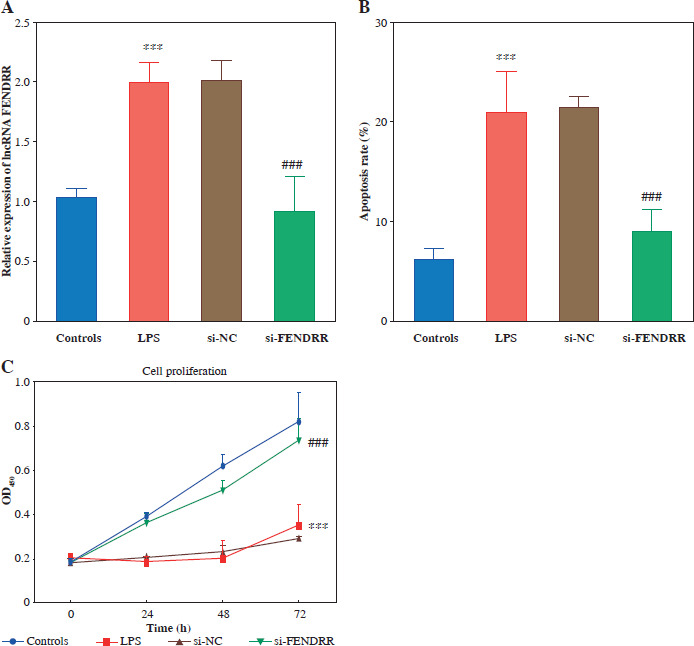
Based on this, we constructed a cell model of sepsis-induced AKI with LPS-stimulated HK-2 to explore the effects of FENDRR on cell function and inflammation during AKI. After LPS treatment, FENDRR significantly increased in the cell model (p < 0.001). The expression of FENDRR was significantly reduced after transfection of si-FENDRR (p < 0.050, Fig. 2A). Subsequently, cell function experiments were performed. Flow cytometry assay showed that LPS treatment significantly increased the apoptosis rate (p < 0.001), whereas silencing of FENDRR reduced the LPS-induced apoptosis rate (p < 0.001, Fig. 2B). Cell viability (24 h, 48 h, and 72 h) was significantly decreased after LPS induction (p < 0.001), and FENDRR downregulation significantly enhanced LPS- induced cell viability (p < 0.001, Fig. 2C).
Fig. 2
Silencing FENDRR reversed the promotion of apoptosis and the suppression of proliferation due to lipopolysaccharides (LPS) treatment in HK-2. A) Expression of FENDRR after transfection. B) Silencing FENDRR reversed the promotion of apoptosis after LPS treatment. C) Silencing FENDRR reversed the suppression of proliferation under LPS treatment. Data are presented as mean ± SD (n = 3). ***p < 0.001 vs. controls, ###p < 0.001 vs. LPS
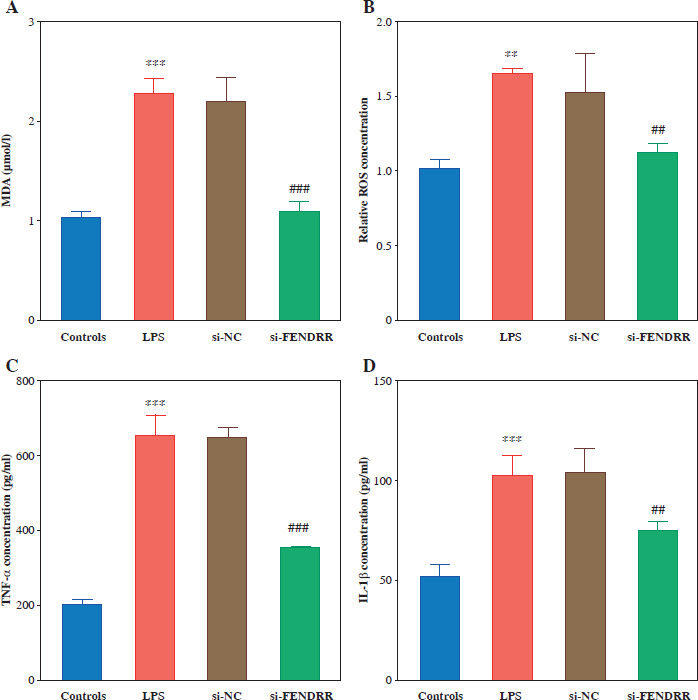
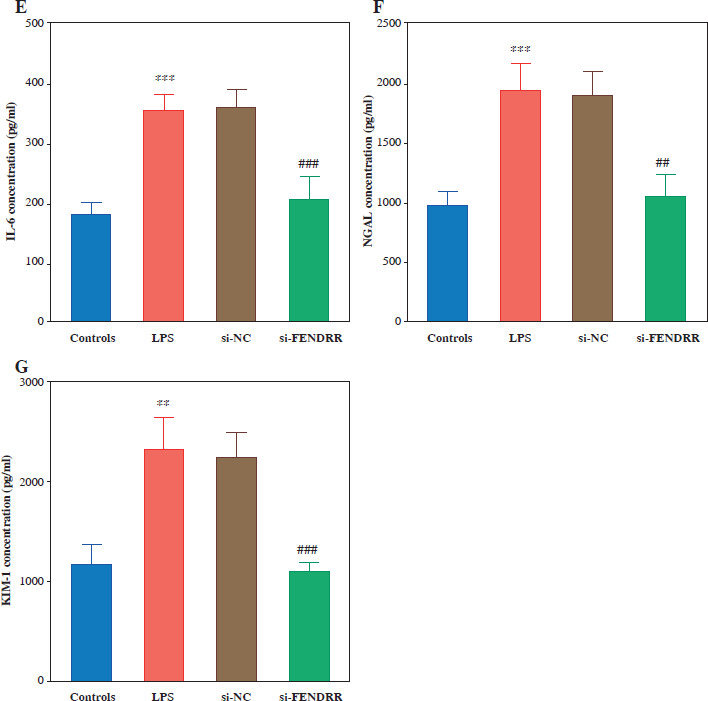
Malondialdehyde and ROS levels were elevated in HK-2 cells after LPS treatment, and silencing of FENDRR reduced LPS-induced MDA and ROS levels in HK-2 cells (p < 0.010, Fig. 3A, B). Silencing FENDRR decreased the levels of inflammatory factors (TNF-α, IL-1β, and IL-6) in the HK-2 cells (p < 0.010, Fig. 3C-E). To strengthen the connection with the in vitro study, the renal tubular damage marker was detected. Down-regulated FENDRR reduced LPS-induced NGAL and KIM-1 levels in the cell model (p < 0.010, Fig. 3F, G).
Fig. 3
Silencing FENDRR reversed the upregulation of inflammatory levels via lipopolysaccharides (LPS) treatment in HK-2. The levels of malondialdehyde (MDA; A), reactive oxygen species (ROS; B), inflammatory factor TNF-α (C), inflammatory factor interleukin (IL)-1β (D). ***p < 0.001 vs. controls, ###p < 0.001 vs. LPS Inflammatory factor IL-6 (E), renal tubular damage marker NGAL (F), and renal tubular damage marker KIM-1 (G) in HK-2 cell model. Data are presented as mean ± SD (n = 3). ***p < 0.001 vs. controls, ###p < 0.001 vs. LPS
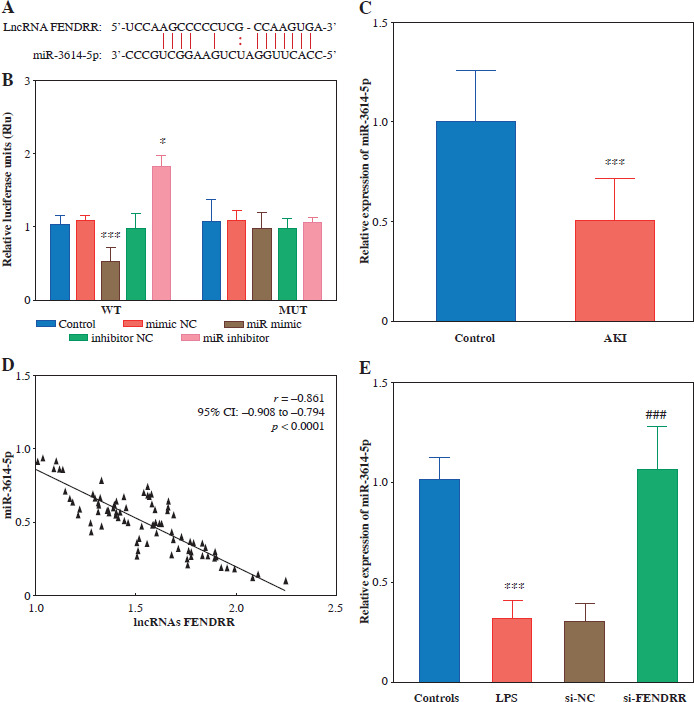
FENDRR negatively regulated miR-3614-5p in LPS-induced HK-2 cells
The target binding sites of FENDRR and miR-3614-5p were predicted using the ENCORI database (Fig. 4A). The relative luciferase activity of FENDRR was significantly down-regulated by miR-3614-5p, as observed in HK-2 cells (p < 0.001). The relative luciferase activity of the co-transfected WT and miR inhibitor groups, on the other hand, increased significantly (p < 0.050). FENDRR relative luciferase activity was not significantly different within the MUT group (p > 0.050, Fig. 4B). To further investigate the relationship between FENDRR and miR-3614-5p, we examined the expression level of miR-3614-5p. The results revealed that miR-3614-5p was significantly downregulated in AKI patients (Fig. 4C). FENDRR and miR-3614-5p were negatively correlated (r = –0.861, p < 0.0001, Fig. 4D). In the LPS-induced cell model, miR-3614-5 was significantly down-regulated (p < 0.001), whereas silencing of FENDRR significantly up-regulated the expression level of miR-3614-5p (p < 0.001, Fig. 4E).
Fig. 4
LncRNA FENDRR binds to miR-3614-5p target. A) Potential binding sites for FENDRR and miR-3614-5p according to ENCORI database. B) The targeting relationship between FENDRR and miR-3614-5p was verified by dualluciferase assay. C) Expression of miR-3614-5p was down-regulated in the serum of AKI patients (n = 85) compared to the control group (n = 90). D) The negative correlation between expression of FENDRR and miR-3614-5p in acute kidney injury (AKI; E). Silencing lncRNA FENDRR reduced miR-3614-5p expression under LPS treatment. Data are presented as mean ± SD (n = 3). ***p < 0.001 vs. controls, ###p < 0.001 vs. LPS
Discussion
Sepsis arises from a dysregulated host response to infection, ultimately leading to life-threatening organ dysfunction [19]. Studies suggested that sepsis-induced AKI is linked to an enhanced systemic inflammatory response. This leads to dysfunctional hemodynamics, causing dilation of the small renal arteries, a reduced glomerular filtration rate, and infiltration of inflammatory cells into the renal parenchyma. These processes result in ischemia, hypoxia, and prolonged kidney underperfusion, further impairing renal function [22, 23]. Most previous studies evaluated the effect of FENDRR on the risk of cancers such as esophageal, colon, and lung cancers [13, 24, 25]. In the present study, FENDRR was found to be abnormally expressed in patients with AKI. ROC analysis has revealed that serum FENDRR possesses significant diagnostic potential for distinguishing between patients with sepsis and those with sepsis-induced AKI. Consequently, FENDRR is emerging as a potential biomarker for sepsis-induced AKI. Logistic analysis revealed that PCT, BUN, SCr, SOFA, lactate, and FENDRR were risk factors for sepsis-induced AKI. PCT is recognized as a marker of acute inflammation and is widely employed in in clinical practice for predicting infectious diseases. Following an infection, serum PCT rises rapidly in a short period under the combined effect of bacterial toxins and inflammatory factors [26]. BUN and SCr are the hallmark indicators of AKI [27]. The SOFA score serves as an effective means to gauge organ function in patients experiencing infectious shock and correlates closely with patient prognosis [28]. APACHE II, known as Acute Physiology and Chronic Health Status Score II, can assess the severity and prognosis of the disease [29]. During acute kidney injury, disruptions in renal glycolysis and gluconeogenesis can significantly disturb lactate metabolism, resulting in altered lactate levels and further impacting the severity and prognosis of AKI [30]. Furthermore, compared to patients without AKI, those who developed AKI had a longer hospitalization stay [31]. In this study, FENDRR was positively correlated with PCT, BUN, SCr, SOFA, APACHE II, lactate, and length of hospital stay. Our finding demonstrated that FENDRR is intricately linked to the severity of sepsis-induced AKI, affirming its role as a biomarker for both the diagnosis and progression of this condition. Additionally, considering the cost aspect, blood biopsy based on qPCR is more cost-effective compared to conventional diagnostic approaches. FENDRR, as a promising biomarker, potentially offers earlier detection and higher sensitivity. It facilitates early detection, real-time monitoring, and assessment of treatment efficacy.
Sepsis disrupts immune homeostasis, activating several signaling pathways, including NF-κB, PI3K, and PKB. Dysregulated inflammation, from early pro-inflammatory factors to late mediators, contributes to the development of AKI and may drive fatal tissue damage and organ dysfunction in late-stage sepsis [4]. Therefore, the inflammatory response is illustrated as the pathogenic mechanism of sepsis-induced AKI. The specific role of FENDRR in sepsis-induced AKI was further investigated by constructing an inflammatory cell model. Previous investigations have indicated that lncRNAs play a pivotal role in sepsis-related AKI. For example, lncRNA XIST modulates cellular inflammation and apoptosis, thereby mitigating sepsis- induced AKI [32]. It has been shown that silencing lncRNA RMRP reduces the production of cellular inflammatory factors [9]. PVT1 regulated NLRP3-mediated pyroptosis in septic AKI in an HK-2 cell model [33]. Lnc-MALAT1 was increased in both the serum of sepsis patients and HK-2 cells injured by LPS [34]. These observations suggest that FENDRR may be involved in the development of sepsis-induced AKI through cellular inflammation and cellular injury. However, there are currently no detailed studies on the specific role of FENDRR in sepsis-induced AKI. In this study, after LPS induction, the apoptosis rate was increased and cell activity was decreased. Conversely, after silencing FENDRR, the apoptosis rate and cell activity of HK-2 cells remained at normal levels after LPS induction. Additionally, silencing of FENDRR significantly alleviated LPS-induced cellular oxidative damage (MDA and ROS content) and down-regulated intracellular inflammatory factor levels (TNF-α, IL-1β, and IL-6). Tubular injury is often an early event in the progression of AKI, with NGAL and KIM-1 recognized as reliable markers of renal tubular damage. The accumulation of these markers was associated with the increased risk of adverse outcomes in kidney dysfunction [35]. Silencing of FENDRR reversed the levels of renal tubular damage markers (NGAL and KIM-1). Based on these results, it is speculated that FENDRR upregulates reactive oxygen species, inflammatory factors, and renal tubular damage, exacerbating the development of AKI.
MiRNAs can modulate unbalanced inflammatory responses by influencing inflammatory factors and inflammatory factor signaling pathways at the post-transcriptional level [17, 36, 37]. In recent years, studies in sepsis have identified a role for numerous miRNAs [38]. For example, lncRNA XIST plays an important role in adaptive immunity by regulating miR-155-5p, which is involved in sepsis [38]. LncRNA RMRP promotes the progression of AKI by upregulating DDX5 in a miR-206-dependent manner [9]. Notable induction of miR-202-5p levels within renal tubular cells in septic AKI in both in vivo and in vitro models, associated with the NF-κB pathway, was reported [39]. To investigate whether FENDRR contributes to sepsis-induced AKI via targeting specific miRNAs, we identified potential binding sites for FENDRR and miR-3614-5p and confirmed the accuracy of the predicted targets by a dual luciferase reporter assay. miR-3614-5p is closely associated with cellular inflammatory responses [18]. In this study, miR-3614-5p was significantly downregulated in AKI patients and negatively correlated with the expression level of FENDRR. Cellular experiments demonstrated that LPS in HK-2 downregulated miR-3614-5, which is consistent with previous reports [18]. Furthermore, miR-3614-5p was significantly upregulated in LPS-induced HK-2 cells after silencing FENDRR. Therefore, it is plausible that miR-3614-5p acts as a target gene of FENDRR in the context of sepsis-induced AKI, modulating cellular inflammation and oxidative damage.
This study has several limitations. For example, SOFA and APACHE II can reflect the prognosis of patients with sepsis-induced AKI, and FENDRR correlates with these two indicators, which suggests that FENDRR may also have potential value as a prognostic marker for sepsis-induced AKI. Further studies are still needed in the future. The effects of FENDRR were only explored in cell experiments, lacking in vivo experimental verification. The investigation did not encompass the potential downstream mRNA targets of the FENDRR/miR-3614-5p axis nor the related signaling pathways. Further studies should consider the ex vivo and complete regulatory axis of FENDRR to understand its mechanism fully. Failure to rigorously consider potential confounding variables that may confound the observed correlations is a limitation of our study. Future studies should aim to conduct more comprehensive data analyses, such as multiple linear regression, to reveal the relationships between variables and help clarify the independent role of FENDRR.
In conclusion, this study suggests that lncRNA FENDRR has potential to predict the risk of AKI in sepsis patients, identify onset and predict severity. LncRNA FENDRR inhibits miR-3614-5p to promote cellular oxidative stress and inflammatory factor levels, exacerbating the cellular injury and therefore boosting sepsis-induced AKI.