Introduction
Gout is a common type of inflammatory arthritis in men caused by hyperuricemia, which can even lead to joint deformities and chronic kidney failure in severe cases [1]. At present, the prevalence of gout in the Chinese Mainland is approximately 1-3%, and its number has exceeded 17 million [2]. Globally, gout affects approximately 1-4% of the population, with higher prevalence in Western countries (e.g., 3.9% in the U.S. and 2.5% in the UK) due to dietary and genetic factors [3-5]. In Europe, gout prevalence ranges from 0.9% to 2.5%, with rising incidence linked to aging populations and metabolic comorbidities [6, 7]. Colchicine and nonsteroidal anti-inflammatory medications are widely used to treat acute gouty arthritis, but their long-term usage is restricted due to unavoidable adverse effects [8]. For chronic management, urate-lowering therapies such as allopurinol (a xanthine oxidase inhibitor) and febuxostat are recommended to prevent recurrent attacks and reduce tophi formation by maintaining serum urate levels below 6 mg/dl [9, 10]. However, urate-lowering therapies require careful dose titration and monitoring due to risks of hypersensitivity reactions (allopurinol) or cardiovascular events (febuxostat) [10, 11]. As a result, new gout treatment options must be developed as soon as possible. Macrophages are one of the first immune cells identified and researched, mediating the entire process of acute gout [12]. Hyperuricemia leads to the formation of monosodium urate (MSU), which may be recognized by the immune system as a danger signal and stimulate macrophage activation, and eventually induce macrophages to produce and release interleukin (IL)-1β and IL-18, causing an inflammatory cascade [13]. Notably, synovial cells play an important regulatory role in macrophage inflammation during gout progression by secreting molecules or cytokines [14]. Therefore, inhibiting macrophage inflammation is a potential therapeutic strategy for gout, and a deeper understanding of the interaction mechanism between synovial cells and macrophages is key to achieving this goal.
The sialyl-Tn (sTn) antigen is a tumor-associated glycan structure that is highly expressed in various diseases and malignancies [15]. Carbohydrate antigen 72-4 (CA72-4) is a common sTn antigen identified by two monoclonal antibodies, CC49 and B72.3, and is a biomarker for multiple malignant tumors, including gastric and breast cancer [16, 17]. Notably, serum CA72-4 is significantly increased in gout [18]. Our previous work also demonstrated that CA72-4 was specifically overexpressed in non-tumor gout patients and can predict flares [19]. In addition, our preliminary research found that CA72-4 was mainly secreted by MSU-induced synovial cells, and secreted CA72-4 could further upregulate transforming growth factor β1 (TGF-β1) in macrophages. However, the mechanism of MSU-induced secretion of CA72-4 by synovial cells is still unclear and deserves further research. As reported, the Tn antigen is formed by core 1 synthase-mediated transfer of galactose to GalNAc Ser/Thr [20]. Core1β3-galactosyltransferase-specific molecular chaperone (Cosmc) is the unique chaperone of T-synthase and is necessary for its functional formation [21]. The increased sTn expression in diseases is mainly related to the loss of Cosmc expression [20, 22, 23]. Nevertheless, whether MSU-induced increase in CA72-4 secretion by synovial cells is caused by the loss of Cosmc expression remains unknown, which deserves further research. DNA methylation modification is the process of adding methyl groups to cytosine or adenine, resulting in decreased gene expression, which is a common epigenetic modification [24]. A previous study showed that CD3/CD28 Dyna beads increased Tn antigen expression in T cells accompanied by Cosmc promoter hypermethylation [25]. In addition, DNA methylation inhibition could restore the epigenetic silencing of Cosmc and reduce Tn antigen expression in human leukocytes [26]. The current study also used bioinformatics to anticipate the presence of several cytosine-phosphate-guanine (CpG) islands in the Cosmc promoter. Therefore, it is speculated that the MSU-induced increase in CA72-4 secretion and the loss of Cosmc expression in synovial cells are related to hypermethylation of the Cosmc promoter.
Long noncoding RNAs (lncRNAs) are a class of transcripts > 200 nucleotides long that lack protein-coding potential but regulate gene expression through diverse mechanisms, including chromatin remodeling, transcriptional interference, and scaffolding of macromolecular complexes [27, 28]. LncRNAs are key epigenetic regulators that modulate gene expression through chromatin remodeling, histone modification, and transcriptional interference [28, 29]. These mechanisms enable lncRNAs to fine-tune inflammatory responses, including NLRP3 inflammasome activation, in diseases such as gout [30, 31]. The dysregulation of lncRNA contributes to the progression of gout. As evidence, Zhang et al. demonstrated that MSU treatment significantly reduced lncRNA-MM2P expression in macrophages, and lncRNA-MM2P overexpression significantly inhibited MSU-induced pro-inflammatory cytokine production in macrophages [32]. In addition, lncRNA HOTAIR knockdown reduced the secretion of inflammatory cytokines in MSU-treated macrophages [31]. A previous study showed that NR-026756 (lncSLED1) was significantly upregulated in peripheral blood mononuclear cells (PMBCs) of gout patients [33]. As revealed by Zhang et al., lncSLED1 is related to macrophage-related immunity responses in post-infarct heart failure [34]. However, the role of lncSLED1 in regulating MSU-induced macrophage inflammation remains unclear.
Our findings revealed that CA72-4 secreted by MSU-induced HFLS inhibited macrophage inflammation, and its expression level was regulated by lncSLED1. Further research found that lncSLED1 regulated CA72-4 expression by elevating the methylation level of the Cosmc promoter. These findings provide a theoretical basis for developing new gout treatment strategies.
Material and methods
Cell culture and treatment
Primary human synovial cells (HFLS) were isolated from the articular cartilage tissue of patients as previously reported [35]. Patients with gout undergoing joint replacement therapy for end-stage gouty arthropathy or secondary osteoarthritis were enrolled. All cases exhibited radiographic evidence of bone erosion, cartilage destruction, and/or tophi deposition. The articular cartilage tissues were collected at Zibo Central Hospital. The informed consent forms were obtained from patients, and the Ethics Committee of Zibo Central Hospital approved the experiment, No. 202211022. The articular cartilage tissues were cut into 25 mm3 pieces, and the pieces were incubated with 0.1% hyaluronidase (Sigma-Aldrich, MO, USA) for 15 min, 0.5% proteinase (Sigma-Aldrich) for 30 min, and 0.2% collagenase (Sigma-Aldrich) for 12 h at 37oC. The isolated HFLS were grown in DMEM (Gibco, MD, USA) containing 10% FBS (Gibco), 1% amphotericin B solution, and 1% L-glutamine with 5% CO2 at 37oC. After corresponding processing, HFLS were cultured in a serum-free DMEM/F12 (Gibco) for 48 h, and the conditional medium (CM) supernatant obtained was collected for further use. ATCC (VA, USA) provided THP-1 cells, and cells were cultured in RPMI-1640 (Gibco) containing 10% FBS with 5% CO2 at 37oC. To induce differentiation of THP-1 cells into macrophages, THP-1 cells were incubated with 100 ng/ml phorbol myristate acetate (PMA; Beyotime, Shanghai, China) for 24 h. For MSU treatment, HFLS and macrophages were incubated with 100 µg/ml MSU crystal (Enzo Life Sciences, NY, USA) for 24 h.
Cell infection
The lentivirus of sh-lncSLED1 (lv-sh-lncSLED1-1, lv-sh-lncSLED1-2, and lv-sh-lncSLED1-3), the lentivirus of sh-Cosmc (lv-sh-Cosmc-1, lv-sh-Cosmc-2, and lv- Cosmc-3), the lentivirus of lncSLED1 overexpression plasmid (lv-lncSLED1), and their negative controls (lv-sh-NC and lv-NC) were obtained from Riobio (Guangdong, China). Cells were infected by lentivirus at MOI = 50 with 40 µl HitransG P virus infection reagent (GeneChem, Shanghai, China) for 16 h. Then, the steadily infected cells were selected by incubation with 2 µg/ml puromycin dihydrochloride (Sigma-Aldrich) for 7 days.
Cell Counting Kit-8 (CCK-8) assay
Cells were plated in the 96-well plates at a density of 2 × 103 cells/well and cultured for 24 h. CCK-8 solution (10 µl; Yeason, Shanghai, China) was added to each well. After incubation at 37oC for 3 h, the absorbance was examined at 450 nm using a microplate reader (Thermo Fisher Scientific, MA, USA).
Flow cytometry
Cells were resuspended in PBS containing 10% FBS at a density of 1 × 106 cells/ml and then stained for 30 min with anti-CA72-4 (Abcam, Cambridge, UK, 1 : 100, ab199002) protected from light. The samples were analyzed by flow cytometry (BD, NJ, USA).
Enzyme-linked immunosorbent assay (ELISA)
The secretion levels of IL-1β, IL-6, IL-10, tumor necrosis factor α (TNF-α), TGF-β1, and CA72-4 were assessed using the human IL-1 β ELISA kit (Abcam, ab214025), the human IL-6 ELISA kit (Abcam, ab178013), the human IL-10 ELISA kit (Abcam, ab185986), the human TNF-α ELISA kit (Abcam, ab181421), the human TGF-β1 ELISA kit (Abcam, ab9758), and the human CA72-4 ELISA kit (Reddot Biotech, Shanghai, China, RD-CA72-4-Hu), respectively. The results were recorded by measuring the OD values at 450 nm.
Methylation-specific PCR (MSP)
DNA methylation of the Cosmc promoter was analyzed by MSP targeting a 115bp CpG island (NC_000023.11:c120632560-120632674). The Tiangen DNA extraction kit (Beijing, China) was used to extract genomic DNA from cells, and the DNA was exposed to bisulfite. MSP of the bisulfite-modified DNA was performed. MSP products were analyzed by gel electrophoresis. Primer sequences for methylated (M-F: 5’-TAATTTTAGTATTGTGGAAGGTCGA-3’; M-R: 5’-ATTACAAATATACGCCACCACGT-3’) and unmethylated (U-F: 5’-TAATTTTAGTATTGTGGAAGGTTGA-3’; U-R: 5’-AATTACAAATATACACCACCACATC-3’) reactions were listed.
Chromatin immunoprecipitation (ChIP) assay
Cells were cross-linked with 1% formaldehyde for 10 min and quenched with glycine for 5 min, Then, the cell lysate was subjected to ultrasound treatment to generate chromatin fragments and incubated overnight with anti-EZH2 (Abcam, 1 : 50, ab307646), anti-H3K27me3 (Abcam, 1 : 30, ab6002), or anti-IgG (Abcam, 1 : 100, ab172730). The chromatin was eluted from the beads and de-crosslinked. The DNA was purified using a centrifugal column and quantified using qPCR.
RNA binding protein immunoprecipitation (RIP) assay
The cell lysates were prepared and incubated with IgG/EZH2-conjugated protein A/G magnetic beads for 1 h. The RNAs were isolated from the beads and analyzed by agarose gel electrophoresis analysis.
Quantitative real-time polymerase chain reaction (qRT-PCR)
TRIzol reagent (Invitrogen, CA, USA) was used to extract bone marrow-derived mesenchymal stem cell (BMSC) RNA. NanoDrop 2000 Spectrophotometers were used to analyze RNA quality. The Superscript III reverse transcriptase kit (Invitrogen) was used to reverse transcribe RNA samples. The data were analyzed using the 2-ΔΔCT method. The primers were as follows (5’-3’):
IL-6 (F): TTCTCTCCGCAAGAGACTTCC
IL-6 (R): TTCTGACAGTGCATCATCGCT
IL-1β (F): TGGGAAACAACAGTGGTCAGG
IL-1β (R): CCATCAGAGGCAAGGAGGAA
TNF-α (F): CCCTCACACTCAGATCATCTTCT
TNF- α (R): GCTACGACGTGGGCTACAG
IL-10 (F): TTGCTGGAGGACTTTAAGGGT
IL-10 (R): CTTGATGTCTGGGTCTTGGTT
TGF- β 1 (F): GAAATTGAGGGCTTTCGCCTTAG
TGF- β 1 (R): GGTAGTGAACCCGTTGATGTCCA
lncSLED1 (F): CAGAGCAGGAGTTGGTGTTGA
lncSLED1 (R): CCGTACTAACTGGGAGCAGC
Cosmc (F): AAGCCGTTCTAGACGCGGGAAA
Cosmc (R): GCTCATGGTGGTGCATTCTA
GAPDH (F): AGGTCGGTGTGAACGGATTTG
GAPDH (R): GGGGTCGTTGATGGCAACA
Western blot
The proteins were extracted from cells using RIPA (Beyotime) and quantified with the BCA kit (Thermo Fisher Scientific). The total protein was isolated by 10% SDS-PAGE and transferred to a Millipore PVDF membrane (MA, USA). The membranes were blocked in 5% non-fat milk and incubated overnight with antibodies against Cosmc (Abcam, 1: 1000, ab229831) and β -actin (Abcam, 1 : 5000, ab8226). After washing with PBS-T, the membranes were then incubated with a secondary antibody (Abcam, 1: 10000, ab7090) for 60 min. After incubation with the secondary antibody, the membranes were visualized using the enhanced chemiluminescence detection kit (Beyotime).
Statistical analysis
Experiments were replicated more than three times. The means ± SD were used to represent the data. The data were analyzed using SPSS 19.0. For comparisons of two groups, an unpaired two-tailed Students’t-test was used. For comparisons of multiple groups, a one-way ANOVA followed by a Tukey post hoc test was employed. The p values less than 0.05 were considered significant.
Results
CA72-4 derived from MSU-treated HFLS inhibited MSU-induced macrophage inflammation
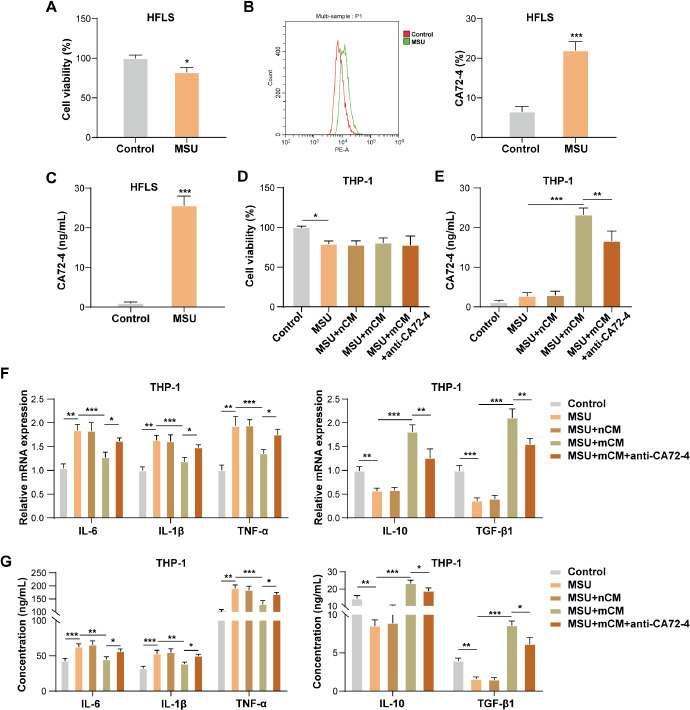
Primary human synovial cells were incubated with MSU, and it was observed that MSU stimulation slightly reduced the vitality of HFLS (Fig. 1A). CA72-4 is a warning molecular marker for gout [18, 19]. Our results showed that MSU treatment significantly elevated the CA72-4 level in HFLS (Fig. 1B, C). To study the role of CA72-4 derived from MSU-treated HFLS in regulating macrophage inflammation during gout progression, the CM was collected from HFLS and MSU-treated HFLS, referred to as HFLS-Non-CM (nCM) and HFLS-MSU-CM (mCM), respectively. THP-1 cells were incubated with 100 ng/ml PMA for 24 h to obtain macrophages, and macrophages were co-treated with CM (nCM or mCM) and anti-CA72-4, then incubated with MSU treatment for 24 h. It was first observed that MSU slightly reduced macrophage vitality, and nCM treatment, mCM treatment, and co-treatment with mCM and anti-CA72-4 had no effect on MSU-treated macrophage vitality (Fig. 1D). In addition, MSU treatment had no significant effect on CA72-4 level in macrophages, and nCM treatment did not affect CA72-4 level in MSU-treated macrophages significantly (Fig. 1E). However, it was also observed that mCM significantly elevated CA72-4 level in MSU-treated macrophages, but this effect of mCM was weakened by anti-CA72-4 treatment (Fig. 1E), further confirming that CA72-4 was derived from MSU-treated HFLS. It was subsequently revealed that MSU stimulation significantly elevated the mRNA and secretion levels of pro-inflammatory cytokines (IL-6, IL-1 β, and TNF-α) and reduced the levels of anti-inflammatory cytokines (IL-10 and TGF- β 1) in macrophages, and these changes induced by MSU were partially eliminated by mCM treatment, while this effect of mCM was weakened by anti-CA72-4 treatment (Fig. 1F, G). Collectively, MSU-treated HFLS inhibited MSU-induced macrophage inflammation by secreting CA72-4.
Fig. 1
CA72-4 derived from MSU-treated HFLS inhibited MSU-induced macrophage inflammation. HFLS were incubated with 100 μg/ml MSU crystal for 24 h. A) The vitality of HFLS was detected by CCK-8 assay. B, C) CA72-4 level in HFLS was examined by flow cytometry and ELISA. THP-1 cells were incubated with 100 ng/ml PMA for 24 h to obtain macrophages, and macrophages were co-treated with CM (nCM or mCM) and anti-CA72-4, and then incubated with MSU treatment for 24 h. D) CCK-8 assay was employed to detect macrophage vitality. E) ELISA was employed to detect CA72-4 levels in macrophages. F, G) the mRNA and secretion levels of IL-6, IL-1β, TNF-α, IL-10, and TGF-β1 in macrophages were assessed using qRT-PCR and ELISA, respectively. The measurement data were presented as mean ± SD. All data were obtained from at least three replicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001LncSLED1 was significantly upregulated in MSU-treated HFLS and was associated with MSU-induced CA72-4 secretion in HFLS
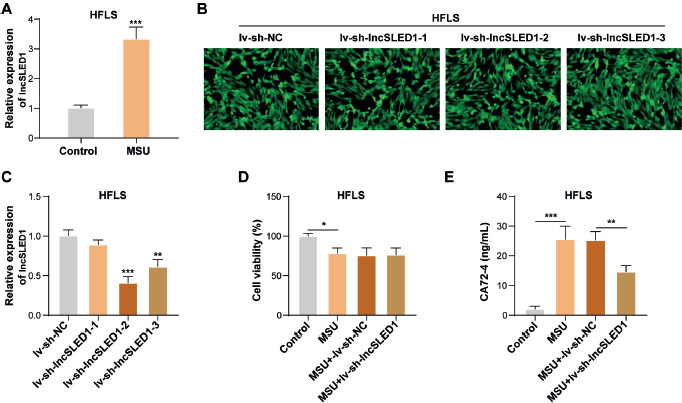
LncSLED1 was previously reported to be significantly upregulated in peripheral blood mononuclear cells (PBMCs) of gout patients [33]. Our results demonstrated that lncSLED1 expression in HFLS was significantly increased by MSU stimulation (Fig. 2A). To study the role of lncSLED1 in regulating MSU-induced CA72-4 secretion in HFLS, lncSLED1 knockdown was induced in MSU-treated HFLS. Observed by fluorescence microscopy, more than 90% of infected target cells expressed green fluorescent protein successfully after lv-sh-NC, lv-sh-lncSLED1-1, lv-sh-lncSLED1-2, or lv-sh-lncSLED1-3 infection (Fig. 2B). It was also observed that lv-sh-lncSLED1-1, lv-sh-lncSLED1-2, and lv-sh-lncSLED1-3 infection significantly reduced lncSLED1 expression in HFLS (Fig. 2C), suggesting that the infection was successful. In addition, the knockdown efficiency of lv-sh-lncSLED1-2 was the highest (Fig. 2C); therefore lv-sh-lncSLED1-2 was selected for subsequent experiments. CCK-8 results subsequently showed that lv-sh-lncSLED1-2 infection had no significant effect on MSU-treated HFLS vitality (Fig. 2D). Additionally, lncSLED1 knockdown ameliorated the MSU-induced increase in CA72-4 secretion in HFLS (Fig. 2E). Taking the evidence together, MSU induced CA72-4 secretion in HFLS by upregulating lncSLED1.
Fig. 2
LncSLED1 was significantly upregulated in MSU-treated HFLS and was associated with MSU-induced CA72-4 secretion in HFLS. A) HFLS were incubated with 100 μg/ml MSU crystal for 24 h, and lncSLED1 expression in cells was examined by qRT-PCR. HFLS were infected with lv-sh-NC, lv-sh-lncSLED1-1, lv-sh-lncSLED1-2, or lv-sh-lncSLED1-3. B) Green fluorescence was observed under fluorescence microscopy. C) qRT-PCR was employed to detect lncSLED1 expression in HFLS. MSU-treated HFLS were infected with lv-sh-NC or lv-sh-lncSLED1. D) CCK-8 assay was performed to examine the vitality of HFLS. E) CA72-4 level in HFLS was assessed by ELISA. The measurement data were presented as mean ± SD. All data were obtained from at least three replicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001
LncSLED1 inhibited MSU-induced macrophage inflammation
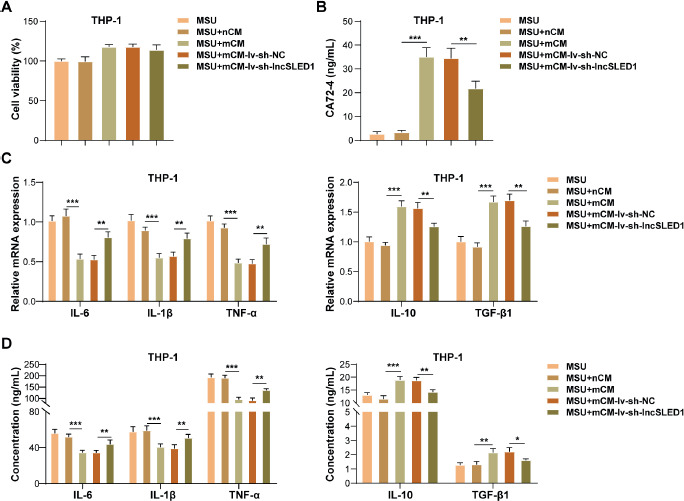
The CM was collected from MSU-treated HFLS after lv-sh-lncSLED1 or lv-NC infection, referred to as mCM-lv-sh-lncSLED1 and mCM-lv-sh-NC, respectively. To study the role of lncSLED1 in regulating MSU-induced macrophage inflammation, macrophages were treated with nCM, mCM, mCM-lv-sh-NC, or mCM-lv-sh-lnc- SLED1, and then incubated with MSU treatment for 24 h. It was first observed that nCM treatment, mCM treatment, mCM-lv-sh-NC treatment, and mCM-lv-sh-lncSLED1 treatment did not affect MSU-treated macrophage vitality (Fig. 3A). In addition, lncSLED1 knockdown in HFLS weakened the promoting effect of mCM on CA72-4 level in MSU-treated macrophages (Fig. 3B). Moreover, lncSLED1 silencing in HFLS partially reversed the inhibitory effect of mCM on the pro-inflammatory cytokines (IL-6, IL-1 β, and TNF-α) and the promoting effect on the levels of anti-inflammatory cytokines (IL-10 and TGF- β 1) in MSU-treated macrophages (Fig. 3C, D). All these results suggested that lncSLED1 knockdown in HFLS reversed the inhibitory effect of mCM on MSU-induced macrophage inflammation.
Fig. 3
LncSLED1 inhibited MSU-induced macrophage inflammation. Macrophages were treated with nCM, mCM, mCM-lv-sh-NC, or mCM-lv-sh-lncSLED1, and then incubated with MSU treatment for 24 h. A) Macrophage vitality was examined using CCK-8 assay. B) ELISA was adopted to determine CA72-4 level in macrophages. C, D) The Mrna and secretion levels of IL-6, IL-1β, TNF-α, IL-10, and TGF-β1 in macrophages were detected by qRT-PCR and ELISA, respectively. The measurement data were presented as mean ± SD. All data were obtained from at least three replicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001
The methylation level of Cosmc promoter was significantly increased in MSU-treated HFLS, which was regulated by lncSLED1
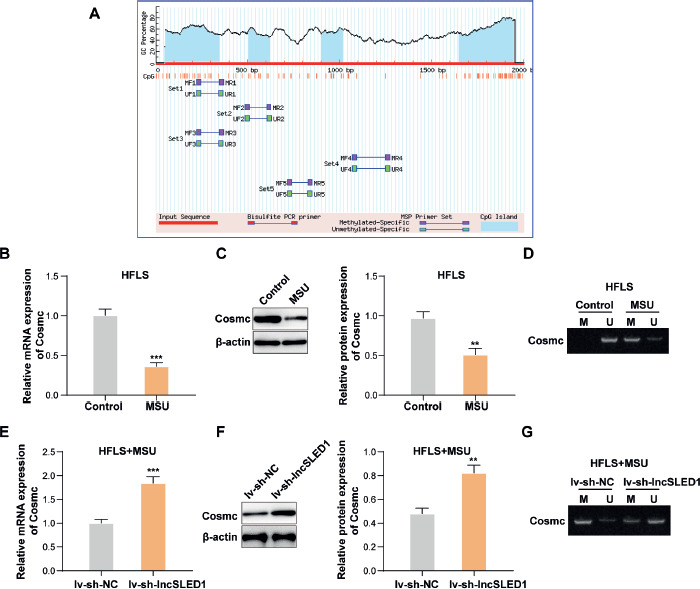
As reported, the abnormal expression of Cosmc is usually caused by abnormal methylation modifications in its promoter region [36]. The abnormal methylation of CpG islands in the promoter is an epigenetic alteration of DNA that is linked to gene silence [37]. As shown in Figure 4A, it was predicted that 3 CpG islands existed in the Cosmc promoter using Methprimer (https://www.methprimer.com/index.html). Our results also showed that MSU treatment significantly reduced Cosmc mRNA and protein levels while elevating the methylation level of the Cosmc promoter in HFLS (Fig. 4B-D). In addition, it was discovered that lncSLED1 knockdown markedly increased Cosmc expression and decreased its methylation level in MSU-treated HFLS (Fig. 4E-G). In summary, lncSLED1 reduced Cosmc expression in MSU-treated HFLS by elevating the methylation level of the Cosmc promoter.
Fig. 4
The methylation level of Cosmc promoter was significantly increased in MSU-treated HFLS, which was regulated by lncSLED1. A) The existence of CpG islands in Cosmc promoter was predicted using Methprimer. B, C) The mRNA and protein levels of Cosmc in HFLS after MSU treatment were determined by qRT-PCR and western blot, respectively. D) MSP was employed to detect the methylation level of the Cosmc promoter in HFLS after MSU. MSU-treated HFLS were infected with lv-sh-NC or lv-sh-lncSLED1. E, F) The mRNA and protein levels of Cosmc in HFLS were determined by qRT-PCR and western blot, respectively. G) The methylation level of the Cosmc promoter in HFLS was detected using MSP. The measurement data were presented as mean ± SD. All data were obtained from at least three replicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001
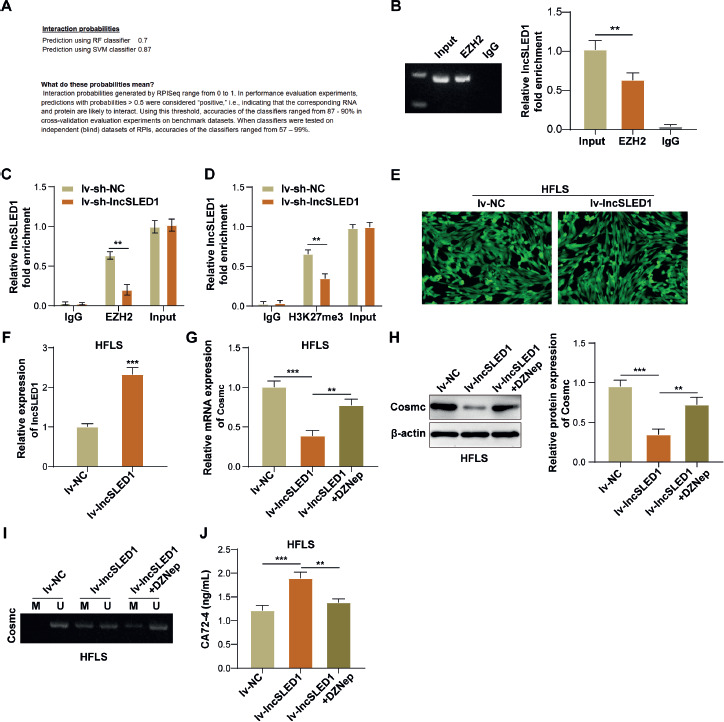
LncSLED1 promoted the methylation level of Cosmc promoter by recruiting EZH2, thereby facilitating CA72-4 secretion in HFLS
It has been reported that lncRNA can regulate the methylation level of the target gene by recruiting EZH2 [34]. EZH2 is a lysine methyltransferase that pre-labels unmethylated CPG islands through activity of its trimethylation histone H3 lysine 27 (H3K27me3) and facilitates DNA methylation in these promoter regions [38]. Herein, it was predicted that lncSLED1 had a high confidence binding relationship with EZH2 using bioinformatics (Fig. 5A). As confirmed by RIP assay, lncSLED1 directly interacted with EZH2 in MSU-treated HFLS (Fig. 5B). ChIP results subsequently demonstrated that the Cosmc promoter directly bound with EZH2/H3K27me3 in MSU-treated HFLS, while this binding relationship was weakened by lncSLED1 knockdown (Fig. 5C, D). HFLS were infected with lv-NC or lv-lncSLED1, and more than 90% of lv-NC/lv-lncSLED1-infected target cells expressed green fluorescent protein successfully (Fig. 5E). Meanwhile, it was observed that lv-lncSLED1 infection significantly elevated lncSLED1 expression in HFLS (Fig. 5F). Furthermore, lncSLED1 overexpression significantly reduced Cosmc expression and increased the methylation level in HFLS, while these effects of lncSLED1 overexpression were partially reversed by DZnep (EZH2 inhibitor) (Fig. 5G-I). It also turned out that the promoting effect of lncSLED1 overexpression on CA72-4 secretion in HFLS was weakened by EZH2 inhibition (Fig. 5J). In conclusion, LncSLED1 facilitated CA72-4 secretion in HFLS by promoting the methylation level of the Cosmc promoter through recruiting EZH2.
Fig. 5
LncSLED1 promoted the methylation level of Cosmc promoter by recruiting EZH2, thereby facilitating CA72-4 secretion in HFLS. A) The potential binding relationship between lncSLED1 and EZH2 was predicted using bioinformatics. B) The interaction between lncSLED1 and EZH2 was analyzed by RIP assay. C, D) ChIP assay was employed to analyze the interactions between Cosmc and EZH2/H3K27me3. HFLS were infected with lv-NC or lv-lncSLED1. E) Green fluorescence was observed under fluorescence microscopy. F) qRT-PCR was employed to detect lncSLED1 expression in HFLS. HFLS were infected with lv-NC or lv-lncSLED1, or co-treated with lv-lncSLED1 and DZNep. G, H) The mRNA and protein levels of Cosmc in HFLS were determined by qRT-PCR and western blot, respectively. I) MSP was performed to determine the methylation level of the Cosmc promoter in HFLS. J) ELISA was adopted to determine CA72-4 level in HFLS. The measurement data were presented as mean ± SD. All data were obtained from at least three replicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001
LncSLED1 inhibited MSU-induced macrophage inflammation by promoting CA72-4 secretion in HFLS through reducing Cosmc expression
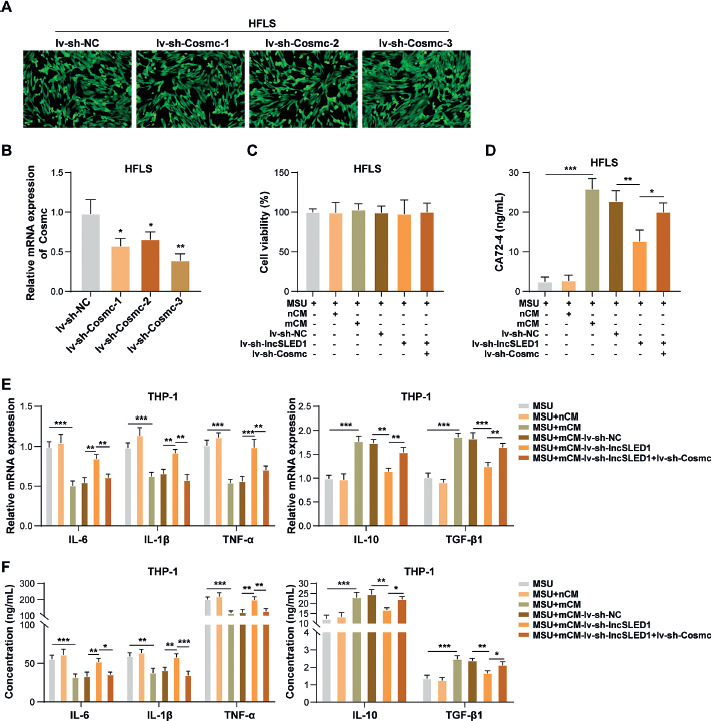
To induce Cosmc knockdown in HFLS, HFLS were infected with lv-sh-NC, lv-sh-Cosmc-1, lv-sh-Cosmc-2, or lv-sh-Cosmc-3. As displayed in Figure 6A, more than 90% of infected target cells expressed green fluorescent protein successfully after lv-sh-NC, lv-sh-Cosmc-1, lv-sh-Cosmc-2, or lv-sh-Cosmc-3 infection. Meanwhile, lv-sh-Cosmc-1, lv-sh-Cosmc-2, and lv-sh-Cosmc-3 infection significantly reduced Cosmc mRNA levels in HFLS (Fig. 6B), suggesting that the infection was successful. In addition, the knockdown efficiency of lv-sh-Cosmc-3 was the highest (Fig. 6B); therefore lv-sh-Cosmc-3 was selected for subsequent experiments. The CM was collected from MSU-treated HFLS after lv-NC, lv-sh-lncSLED1, lv-NC, or lv-sh-lncSLED1 and lv-sh-Cosmc infection; these are hereafter referred to as mCM-lv-sh-NC, mCM-lv-sh-lncSLED1, and mCM-lv-sh-lncSLED1 + lv-sh-Cosmc, respectively. To study the interaction between lncSLED1 and Cosmc in regulating MSU-induced macrophage inflammation, macrophages were treated with nCM, mCM, mCM-lv-sh-NC, mCM-lv-sh-lncSLED1, or mCM-lv-sh-lncSLED1 + lv-sh-Cosmc, and then incubated with MSU treatment for 24 h. It was first observed that mCM-lv-sh-lncSLED1 + lv-sh-Cosmc had no significant effect on MSU-treated macrophage vitality (Fig. 6C). In addition, Cosmc knockdown in HFLS reversed the alleviating effect of lncSLED1 silencing on mCM-induced increase in CA72-4 level in MSU-treated macrophages (Fig. 6D). Moreover, Cosmc silencing in HFLS reversed the weakening effect of lncSLED1 downregulation on mCM-induced inhibition on MSU-induced macrophage inflammation (Fig. 6E, F). In conclusion, lncSLED1 promoted CA72-4 secretion in HFLS through reducing Cosmc expression, thereby inhibiting MSU-induced macrophage inflammation.
Fig. 6
LncSLED1 inhibited MSU-induced macrophage inflammation by promoting CA72-4 secretion in HFLS by reducing Cosmc expression. HFLS were infected with lv-sh-NC, lv-sh-Cosmc-1, lv-sh-Cosmc-2, or lv-sh-Cosmc-3. A) Green fluorescence was observed under fluorescence microscopy. B) Cosmc mRNA level in HFLS was determined using qRTPCR. Macrophages were treated with nCM, mCM, mCM-lv-sh-NC, mCM-lv-sh-lncSLED1, or mCM-lv-sh-lncSLED1 + lv-sh-Cosmc, and then incubated with MSU treatment for 24 h. C) CCK-8 assay was employed to assess macrophage vitality. D) CA72-4 level in macrophages was measured by ELISA. E, F) The mRNA and secretion levels of IL-6, IL-1β, TNF-α, IL-10, and TGF-β1 in macrophages were detected by qRT-PCR and ELISA, respectively. The measurement data were presented as mean ± SD. All data were obtained from at least three replicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Often associated with severe pain and joint swelling, gout is a common inflammatory arthritis that can seriously affect quality of life and lead to disability. The core pathogenesis of gout is the activation of innate immune cells, especially macrophages, by MSU crystals, which trigger inflammatory responses [39]. Therefore, inhibiting MSU-induced macrophage inflammation is a potential treatment strategy for gout. Our preliminary research found that CA72-4 secreted by HFLS could inhibit MSU-induced macrophage inflammation, but the mechanism regulating CA72-4 secretion in HFLS remains unclear. Our primary findings indicated that lncSLED1 promoted CA72-4 secretion in HFLS by increasing the methylation level of the Cosmc promoter through recruiting EZH2, thereby relieving MSU-induced macrophage inflammation.
CA72-4 is a glycoprotein tumor marker [16, 17]. Recently, it has been widely reported that serum CA72-4 is elevated in gout [18, 19]. Our results showed that MSU stimulation significantly elevated CA72-4 level in HFLS. As reported, increased production of s-Tns in rheumatoid arthritis is related to synovial inflammation [40]. In addition, tumor-associated sTn antigen can promote macrophage M2 polarization and anti-inflammatory cytokine secretion in macrophages in intratumoral microenvironments [41]. However, as a common sTn antigen, the role of CA72-4 in regulating macrophage inflammation during gout remains unclear. Our findings revealed that the CM from MSU-treated HFLS (mCM) could inhibit MSU-induced macrophage inflammation, while this effect of mCM was weakened by anti-CA72-4 treatment. All these results suggested that MSU stimulated HFLS to secrete CA72-4, and CA72-4 derived from HFLS could inhibit MSU-induced macrophage inflammation.
LncRNA has also recently become a new research hotspot in gout. As evidence, lncRNA H19 knockdown inhibited inflammatory responses in gouty arthritis [42]. Recently, a new lncRNA (lncSLED1) has entered our field of vision due to its prominent role in inflammatory diseases. For instance, lncSLED1 expression was markedly increased in PBMCs of systemic lupus erythematosus patients [43]. In addition, lncSLED1 upregulation was related to the secretion of pro-inflammatory cytokine (IL-1β) in macrophages during the development of post-infarct heart failure [34]. Notably, a previous study demonstrated that lncSLED1 was one of the lncRNAs significantly upregulated in PBMCs of patients with primary gout [33]. Herein, our results demonstrated that lncSLED1 in HFLS was significantly increased by MSU stimulation, and its knockdown inhibited MSU-induced CA72-4 secretion in HFLS. In addition, lncSLED1 knockdown in HFLS reversed the inhibitory effect of mCM on MSU-induced macrophage inflammation. Collectively, lncSLED1 inhibited MSU-induced macrophage inflammation by promoting CA72-4 secretion in MSU-treated HFLS.
Cosmc participates in the synthesis of Tn antigen [23]. The loss of Cosmc or active T-synthase can increase the expression of sTn antigen [22]. Our results showed that lncSLED1 promoted CA72-4 secretion in MSU-treated HFLS by reducing Cosmc expression. As widely reported, the abnormal expression of Cosmc under pathological conditions is related to DNA methylation in its promoter region [36]. In addition, demethylation of the Cosmc promoter could restore its inhibitory effect on sTn antigen expression [44]. Our findings revealed that lncSLED1 reduced Cosmc expression in MSU-treated HFLS by increasing the methylation level of the Cosmc promoter. The mechanism involved was subsequently studied. As reported, lncRNA can recruit DNA methyltransferase (DNMT) into the promoter of the target gene and regulate the methylation level of the target gene promoter by binding to EZH2 [34]. LncRNAs can regulate the methylation level of downstream targets by recruiting EZH2 [34, 45]. EZH2 is a histone methyltransferase which works as an enzymatic catalytic component in polycomb repressive complex 2 (PRC2) and regulates gene expression by methylating H3K27 in nucleosomes [46]. In addition, EZH2 interacts with DNMT to methylate CpG islands of target genes, inhibiting gene transcription [47]. A similar mechanism was observed in HFLS. Our results demonstrated that lncSLED1 promoted the methylation level of the Cosmc promoter in MSU-treated HFLS by recruiting EZH2. Mechanistically, lncSLED1 promoted CA72-4 secretion in MSU-treated HFLS by reducing Cosmc expression and increasing the methylation level of the Cosmc promoter through recruiting EZH2.
A key finding of this study is the negative correlation between elevated CA72-4 levels and macrophage-derived inflammatory cytokines (e.g., IL-6, TNF-α), suggesting its potential role as an endogenous compensatory mechanism to dampen excessive inflammation during gout flares. Similar anti-inflammatory feedback loops have been documented in other chronic inflammatory diseases. For example, in rheumatoid arthritis, elevated levels of IL-1Ra are associated with suppressed joint inflammation, representing a similar feedback loop [48]. Our previous work demonstrated that high serum CA72-4 predicts reduced gout flare risk [19], aligning with its putative protective function. Therapeutically, exogenous administration of CA72.4 could theoretically dampen joint inflammation and decrease flare frequency. Notably, colchicine – a cornerstone gout therapy – was reported to upregulate serum CA72-4 levels in gout patients [49], suggesting that its anti-inflammatory effects may partially rely on CA72.4 induction. This aligns with our observation of CA72.4 as an endogenous brake on inflammation. Furthermore, combining CA72.4 with existing therapies (e.g., colchicine or anti-IL-1 biologics) might yield synergistic effects, as seen with dual cytokine blockade in rheumatoid arthritis [50]. However, conclusive evidence for its clinical efficacy remains lacking, particularly regarding optimal delivery methods and potential off-target effects. Future studies should prioritize randomized controlled trials to evaluate CA72.4-based interventions, coupled with mechanistic studies to clarify its signaling pathways.
In addition, our current study relies primarily on the MSU-stimulated HFLS cell model, an immortalized synovial fibroblast line that may differ from primary synovial fibroblasts in epigenetic regulation and secretory profiles, potentially influencing the physiological relevance of CA72-4 secretion regulation. However, we selected the HFLS model based on its well-established role in gout synovitis research [51-53], particularly for studying MSU crystal-induced pro-inflammatory factor secretion. Similarly, while THP-1-derived macrophages have been widely used in gout-related inflammation studies [54-56], we also acknowledge that primary cell and in vivo validation will be critical for translational relevance. Nevertheless, these standardized cellular systems provided a controlled and reproducible platform for dissecting the lncSLED1-CA72-4 regulatory axis at the molecular level. Additionally, regarding the absence of in vivo animal model validation, we acknowledge this as an important limitation. Given the scope and timeline of the current study, we prioritized cellular-level mechanistic investigations to lay the foundation for future translational work. We are committed to conducting animal experiments in subsequent studies to confirm our current results and delve deeper into the biological effects.
Taken together, lncSLED1 upregulation inhibited MSU-induced macrophage inflammation by promoting CA72-4 secretion in MSU-treated HFLS by elevating the methylation level of the Cosmc promoter via recruiting EZH2. In the current research, we did not validate our conclusion in vivo, which is the limitation of our research. Our research provides a reference for further research on the pathogenesis of gout.