Introduction
Ischemic stroke (IS) is a neurological disorder associated with high mortality and morbidity. The current predominant therapeutic strategy remains the restoration of cerebral blood supply via revascularization. However, blood reperfusion benefits only a small subset of stroke patients due to the narrow therapeutic window and the potential for reperfusion-induced inflammation, which increases the risk of neuronal damage and poor prognosis [1]. The resident cerebral immune cells presenting throughout the central nervous system, microglia, play a vital role in post-injury inflammatory response and brain damage. Microglia can be activated and release diverse proinflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor α (TNF-α), to attract peripheral neutrophils, macrophages, and T cells, to infiltrate around the infarcted cortex and cause further neurotoxicity [2, 3]. Therefore, inhibiting microglia activation and neuroinflammatory responses is suggested as a promising therapeutic strategy for IS despite the unclear mechanism of ischemia/reperfusion (I/R) microglia activation.
Autophagy is documented as a progression of lysosome-mediated self-degradation that eliminates damaged or aged proteins and organelles to maintain intracellular homeostasis [4]. In neurons, autophagy can have dual effects: it may protect cells by clearing damaged components, or contribute to cell death under pathological conditions. Moderate autophagy benefits maintain intracellular ROS homeostasis, triggering protection of brain tissue and cells against cerebral I/R injury by mediating intracellular ROS levels. In contrast, excessive autophagy could be activated by overexpression of ROS that aggravates neuronal cell death and tissue damage [5-7]. Additionally, macrophage polarization has controversial effects in cerebral I/R injury. The M1-like phenotype is associated with uncontrolled neuroinflammation, often observed in neurodegenerative diseases. In contrast, the M2-like phenotype promotes inflammation resolution and tissue repair [8]. Neuroprotection via reduction of cerebral cell death, achieved by inhibiting excessive autophagy and stimulating macrophage polarization from M1 to M2 against brain I/R injury, therefore, is a potential strategy in treating IS [9]. IL-33 is a member of the IL-1 family of cytokines involved in the pathogenesis of central nervous system (CNS) disorders such as neurodegenerative diseases, stroke, and infectious diseases. It has been identified as an essential regulator of neuroinflammation [10]. A study suggested that IL-33/ST2 signaling is crucial for microglia and brain endothelial cell activation [11]. Moreover, IL-33 can stimulate ROS generation, improving autophagic levels by producing IL-13 [12].
Nevertheless, the mechanism of IL-33 in promoting autophagy of post-oxygen-glucose-deprivation/reoxygenation microglial via macrophage polarization remains a mystery. This study aimed to preliminarily explore the function of IL-33 in IS by investigating the relationship among IL-33, macrophage polarization, and autophagy, which could provide new therapeutic insights and clinical targets.
Material and methods
Animals
Two-day-old (P2) newborn C57BL/6 mice were provided by ZHBY Biotech Co. Ltd. (Jiangxi, China). All mice were treated in strict accordance with the guidelines of the Ethics Committee of Fujian Medical University, and all animal experiments were approved by the Ethics Committee of Fujian Medical University (IACUC FJMU 2023-Y-0438). Efforts were made to reduce the number of mice used and their suffering.
Primary brain microglia and bone marrow-derived macrophage (BMDM) isolation
Primary brain microglia were isolated from P2 newborn C57BL/6 mice. In brief, the neonatal mice cerebral cortices without blood vessels and meninges were rinsed repeatedly for blood stain removal with cold Hank’s solution brain, followed by digestion with 0.25% trypsin for 10 minutes. The precipitate was incubated in DMEM/F12 with 10% FBS in an incubator at 37°C in 5% CO2 and 95% air to halt digestion. The filtered mixed glial cells were centrifuged at 1000 r/min for 5 min to obtain sediment, which was afterward resuspended in DMEM/F12 with 20% FBS. Cells were seeded into 75 cm2 flasks at a density of 3 × 105 cells/cm2 and cultured for 9-11 days. When the cells reached confluence and were maintained for 3 days (the 11th day), the culture medium was switched to DMEM/F12 with 10% FBS and the culture flasks were placed in a shaking incubator for 12 hours to collect the floating cells, which represented the pure primary microglia.
Mouse bone marrow cells isolated by flushing the marrow space of the femur and tibia with 10% complete culture medium in 5-week-old mice were dispersed into a single-cell suspension. After adding erythrocyte lysate for 10 minutes and discarding the supernatant, the floating cells resuspended by medium were treated with 40 ng/ml GM-CSF for 8 days and cultured in the incubator at 37oC in 5% CO2.
Transient cell transfection with si-IL-33/si-NC
When the density of microglia reached 70% confluence, the culture medium was replaced with 1 ml of serum-free medium. The lyophilized siRNA powder (si-IL-33 or si-NC, Jiangxi Zhonghong Boyuan Biotechnology Co., Ltd) was reconstituted in DEPC-treated water at a concentration of 125 µl per OD unit.
For transfection, 5 µl of Lipofectamine 3000 (L3000015, Invitrogen) and 12.5 µl of reconstituted siRNA were separately diluted in 125 µl of Opti-MEM medium (Gibco), incubated for 5 minutes at room temperature, then combined and incubated for an additional 15 minutes to allow complex formation. The transfection mixture was then added dropwise to the cells. After 4 hours of transfection, the medium was replaced with complete culture medium containing 20% serum, and cells were incubated for 48 hours. Transfection efficiency was evaluated by Western blot and qRT-PCR.
Microglia exposure to OGD/R injury
For imitating oxygen–glucose deprivation, the si-IL-33 (or not) microglia were cultured with the sugar-free medium in an incubator with 94% N2, 5% CO2 and 1% O2 for 2 hours. To induce reperfusion injury, the medium of cells was switched to the regular medium in an incubator at 37oC in 5% CO2 and 95% air for another 24 hours.
Macrophage polarization and macrophage – OGD/R microglia co-cultures
The direct co-culture system was applied in this experiment. The treated microglia and M1 (M2) macrophages were mixed in a ratio of 1 : 2 directly and incubated for 24 hours. The supernatant of the co-culture system was collected for ELISA and WB assay, and the remaining cells were used for immunofluorescence assay.
Quantitative real-time RT-PCR (qRT-PCR) assay
The transcriptional level of IL-33 was detected by qRT-PCR assay for verifying transfection efficiency. Total RNA was extracted from cells using an Ultrapure RNA Kit (CW0581M, CWBIO) and Trizon Reagent (CW0580S, CWBIO). Samples were then reverse-transcribed to cDNA from RNA by HiScript II Q RT SuperMix for qPCR (R223-01, Vazyme). cDNA was amplified by ChamQ Universal SYBR qPCR Master Mix (Q711-02, Vazyme) on a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories Inc.). All procedures followed the manufacturer’s instructions. β-actin was used as an internal reference for normalization, and the relative expression was calculated according to the 2-ΔΔCt method. The primers for IL-33 and β-actin, synthesized by General Biosystems (Anhui) Co., are presented in Table 1.
Western blot
Antibodies against IL-33, STAT1, IRF5, IRF4, CEBP-β, Beclin-1, P62 (1 : 500, AF6300/DF6283/DF6198/AF7747/AF5128/AF5384, Affinity Biosciences Group Ltd.), PPAR-γ, STAT6, IRF8, STAT3, ULK1 (1 : 2000, 16643-1-AP/66717-1-Ig/60199-1-lg/20986-1-ap, Proteintech Group, Inc) and the internal control β-actin (1 : 2000, TA-09, Beijing Zhong Shan Golden Bridge Biological Technology Co., Ltd) were applied. The cells were lysed with a Cell Lysis kit (C1053, Applygen Technologies Inc.) before the cells were placed on ice for 20 minutes. The total proteins were extracted by centrifugation at 12,000 r/min for 10 minutes, and the supernatant containing proteins was retained for subsequent assays. The total proteins were reserved at –20oC. A BCA kit (E-BC-K318-M, Elabscience) was used for verifying protein concentration. Equal amounts of extracted protein were loaded in 10% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) and separated by gel electrophoresis under constant flow at 300 mA for 1.5 hours. The target proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore). The membranes were incubated with primary antibodies at 4°C overnight before incubation with 5% non-fat milk to prevent non-specific binding. After rinsing with TBST, membranes were incubated with diluted horseradish peroxidase (HRP)-conjugated secondary antibodies (1 : 2000, ZB-2305, Beijing Zhong Shan Golden Bridge Biological Technology Co., Ltd) for 2 h the next day. The densities of the binding bands were visualized by the Ultra High Sensitivity Chemiluminescent Imaging System and quantified with the Image J software (National Institutes of Health, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay (ELISA)
The expression of interferon γ (IFN-γ), IL-10, and TGF-β in cell supernatants was measured using ELISA kits (Jiangsu Meimian Industrial Co., Ltd). In accordance with the manufacturer’s instructions, all samples, including standards, were run in triplicate. Linear regression equations for standard curves were calculated with standard concentrations and optical density (OD) 450. The actual sample concentration was obtained by substituting the OD value of the sample into an equation and multiplying it by the dilution factor.
Multiple immunofluorescence assay
All samples were fixed with 4% parapformaldehyde for 30 minutes after rinsing with PBS 3 times. The cells were blocked with 5% BSA at 37°C for 30 minutes. Without washing, cells were incubated overnight at 4°C with primary antibodies against CD16+CD32 (1 : 200, Abcam Trading (Shanghai) Co., Ltd.) and CD206 (1 : 200, Affinity Biosciences Group Ltd.). The next day, cells were washed three times with PBS and incubated with ABflo® 488-conjugated Goat anti-Rabbit IgG (H+L) (1 : 200, AS053, ABclonal) for 30 minutes at 37°C. After three PBS washes, cells were blocked again with 5% BSA at 37°C for 30 minutes. Without washing, cells were incubated with anti- Iba1 primary antibody (1 : 200, Proteintech Group, Inc.) at 37°C for 3 hours, followed by three PBS washes. Cells were then incubated with Cy3-conjugated secondary antibody (1 : 200, AS007, ABclonal) for 30 minutes at 37°C for 30 minutes in the dark. The slides were counterstained with nuclear dye DAPI (Jiangsu KeyGEN BioTECH Co., Ltd.) and observed using a fluorescent microscope.
Statistical analysis
All experiments were performed at least in triplicate. All the data are presented as the mean ± standard error of the mean (SEM). Statistical differences between groups were evaluated using an independent samples t-test, while statistical differences among groups were analyzed by one-way ANOVA. A p-value < 0.05 was considered statistically significant. GraphPad 9.0 software was used for statistical analysis and graphing.
Results
Effect of IL-33 deficiency on macrophage polarization and the inflammatory phenotype of OGD/R microglia
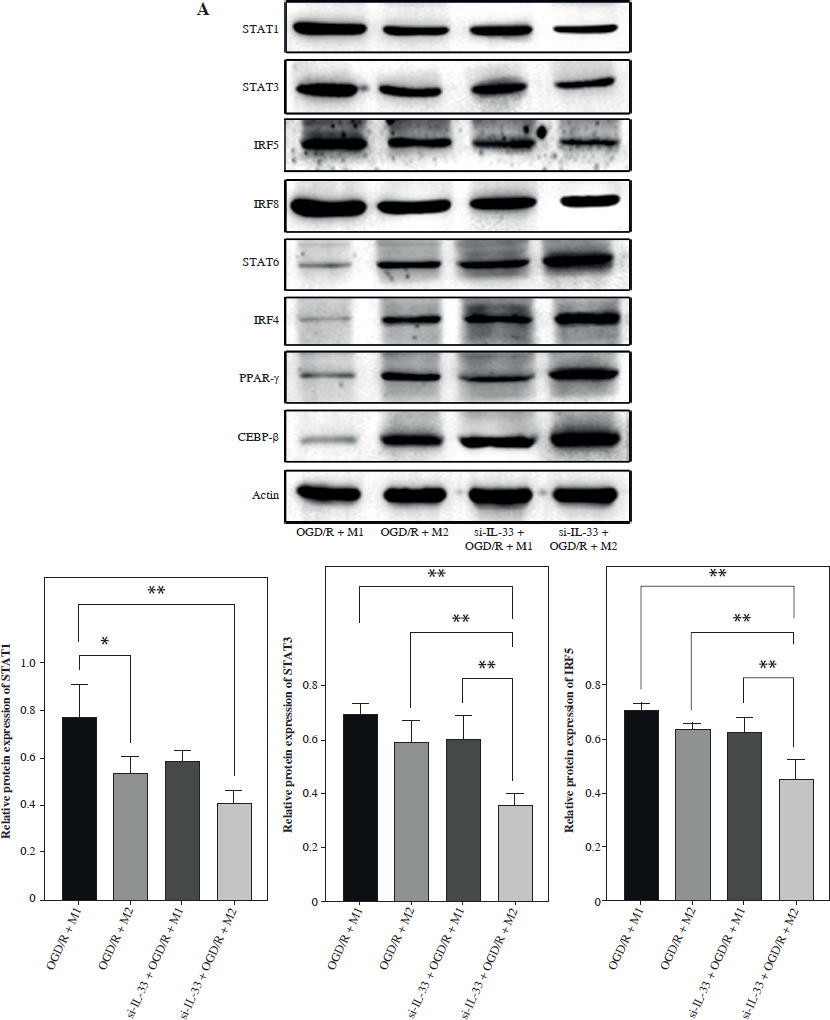
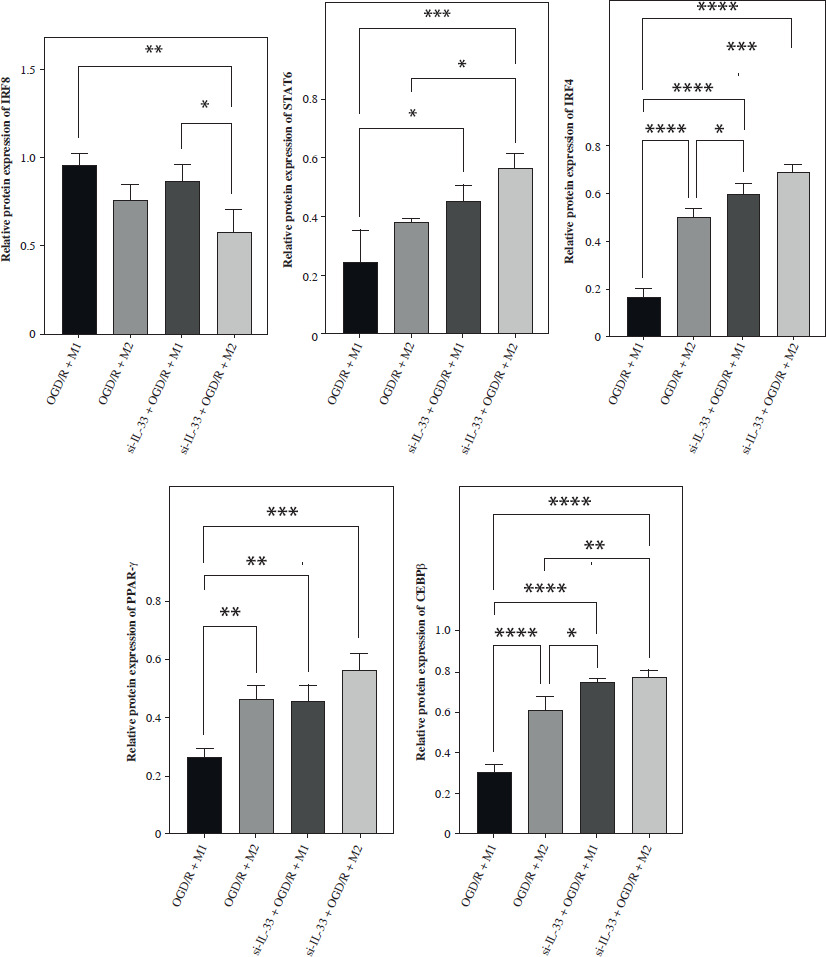
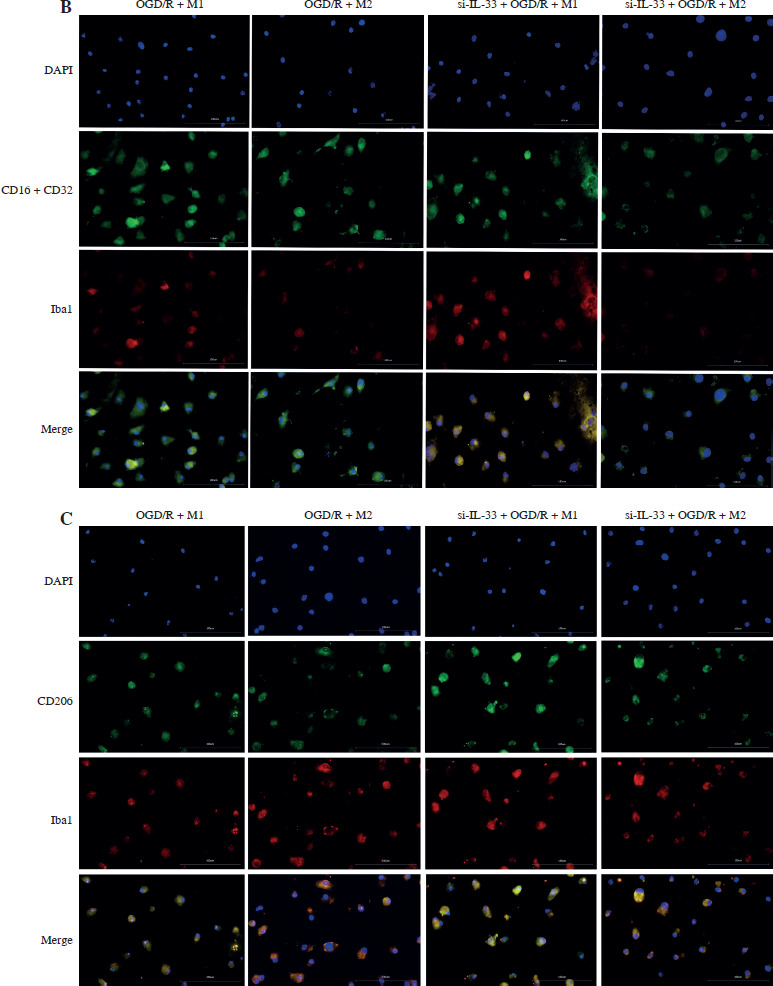
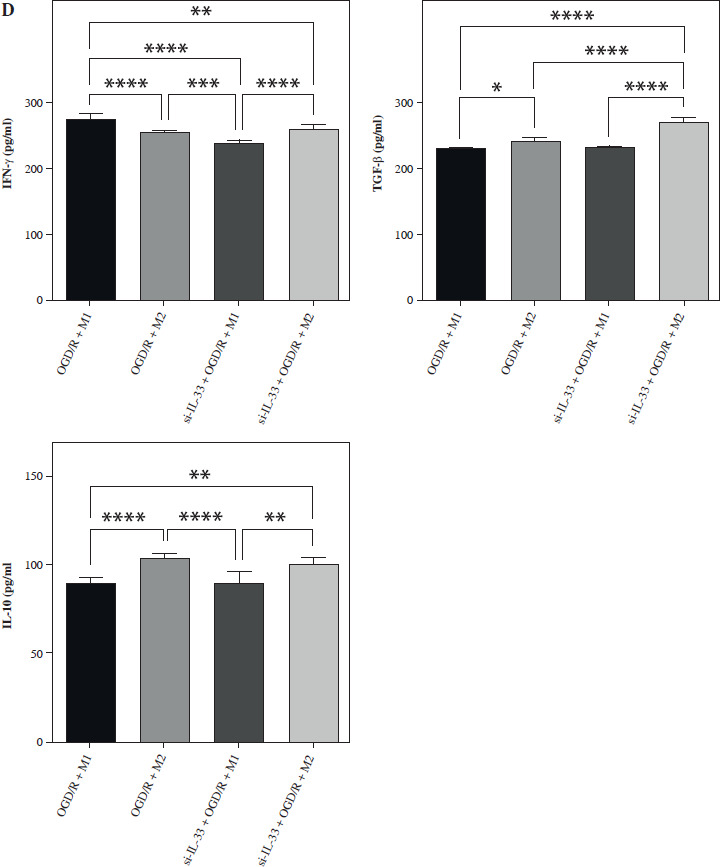
To elucidate the involvement of IL-33 in cerebral I/R, the effect of IL-33 on the relevant cytokines of OGD/R microglia was assessed. The efficiency of the IL-33 interference vector effect was validated by qPCR and WB. To assess changes in M1 and M2 macrophage polarization, intracellular markers of M1 (STAT1, STAT3, IRF5, IRF8) and M2 (STAT6, PPAR-γ, IRF4, CEBP-β) were measured by Western blot (WB) (Fig. 1A). The results from M2/si-IL-33 microglia co-cultures with OGD/R conditions showed that the expression levels of STAT6, PPAR-γ, IRF4, and CEBP-β were significantly increased (p < 0.05), while those of STAT1, IRF8, STAT3, and IRF5 were significantly reduced (p < 0.05). IL-33 knockdown markedly reduced the pro-inflammatory phenotype. The co-expression between M1-type macrophage marker CD16+CD32 and microglial cell marker Iba1 and the co-expression between M2-type macrophage marker CD206 and microglial cell marker Iba1 were detected via immunofluorescence assay (Fig. 1B, C). Compared with M1/si-IL-33 microglia co-cultures, CD206 expression was higher in M2/si-IL-33 microglia co-cultures. ELISA results showed that the M2-type extracellular markers IL-10 and TGF-β were significantly increased (p < 0.05), whereas the M1-type marker IFN-γ was significantly decreased (p < 0.05) in M2/si-IL-33 co-cultures compared with M1/si-IL-33 co-cultures (Fig. 1D).
Fig. 1
Effect of IL-33 deficiency on macrophage polarization and the inflammatory phenotype of OGD/R microglia. A) Expression of M1-type intracellular markers and M2-type intracellular markers was detected by WB. *p < 0.05, **p < 0.01. All experiments were performed in triplicate A) Expression of M1-type intracellular markers and M2-type intracellular markers was detected by WB. *p < 0.05, **p < 0.01, ***p < 0.001 , ****p < 0.0001. All experiments were performed in triplicate B, C) Immunofluorescence detection of M1-type macrophage markers and co-expression of M2-type macrophage markers with microglia markers D) Expression of M1-type extracellular markers and M2-type extracellular markers in cell supernatants by ELISA assay. Compared to the OGD/R + M1 group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All experiments were performed in triplicate
Effect of IL-33 deficiency and macrophage polarization on autophagy of OGD/R microglia
Autophagy, a critical cellular process for the degradation and recycling of damaged organelles and proteins, has been shown to be upregulated in models of oxygen-glucose deprivation (OGD), a common in vitro model for ischemia-reperfusion injury. The expression of autophagy-related proteins such as Beclin-1 and Unc-51-like kinase 1 (ULK1) increases under OGD conditions, while P62, an autophagy adaptor protein, typically shows an inverse relationship with autophagic activity. Elevated Beclin-1 and ULK1 levels indicate enhanced autophagic flux, as Beclin-1 is integral to autophagosome formation, while ULK1 is a key initiator of autophagy signaling pathways. Moreover, P62 is known to accumulate when autophagy is inhibited, serving as a marker for impaired autophagy, while its degradation reflects effective autophagic processes.
In this study, WB analysis was performed to examine the expression of Beclin-1, P62, and ULK1 in the OGD/R model (Fig. 2). Compared to the OGD/R model group, co-culture with M1 macrophages significantly increased the expression of Beclin-1 and ULK1, while reducing P62 expression. The activation of autophagy by M1 macrophages may play a role in facilitating cellular survival under conditions of ischemic stress by removing damaged organelles and proteins [6].
Fig. 2
Effect of IL-33 deficiency and macrophage polarization on autophagy of OGD/R microglia. Expression of autophagy signaling pathway proteins Beclin-1, p62, and ULK1 in cells. Compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All experiments were performed in triplicate
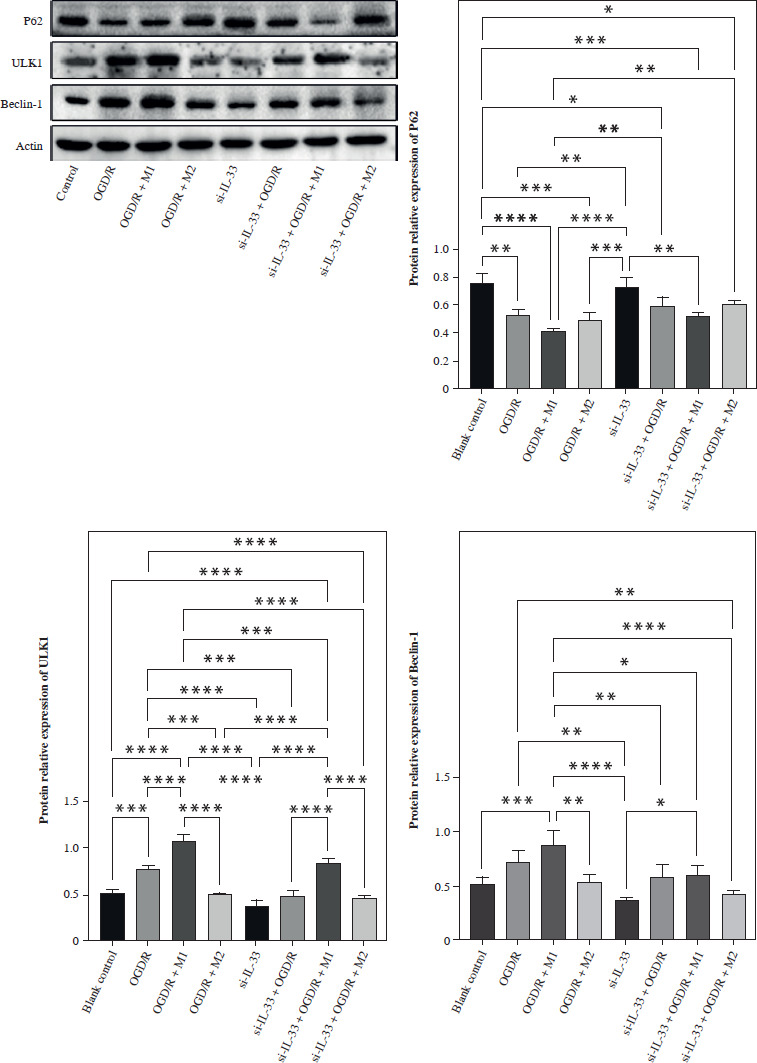
Conversely, co-culture with M2 macrophages and IL-33 deficiency both resulted in a reduction in Beclin-1 and ULK1 expression, alongside an increase in P62 levels, suggesting that M2 macrophages may suppress autophagy. Additionally, the absence of IL-33, an alarmin that has been shown to promote autophagy under certain pathological conditions, further attenuates autophagy signaling, indicating its possible role in regulating the balance between inflammation and autophagy. The increase in P62 levels in these conditions suggests that autophagic degradation is impaired, leading to the accumulation of cellular debris and potentially exacerbating ischemia-reperfusion injury.
In the context of ischemic injury, autophagy serves a dual role: while moderate autophagy can protect cells by removing damaged components, excessive or dysregulated autophagy can lead to autophagic cell death, contributing to tissue damage. Thus, the differential modulation of autophagy by M1 and M2 macrophages, as well as the influence of IL-33, highlights the complex interplay between macrophage polarization, inflammatory responses, and autophagic activity in ischemic injury models.
Discussion
Stroke is a leading cause of mortality and disability worldwide, with ischemic stroke accounting for about 80% of all strokes [13]. Microglial inflammation has emerged as a subject of interest in ischemic stroke therapy. As immune cells of the central nervous system, microglia exhibit a certain degree of protection against ischemic brain injury. However, microglia are incapable of repairing the damage from inflammation [14]. Some studies have suggested that the M2 macrophage plays a neuroprotective role by promoting brain tissue repair, fragment phagocytosis, and regenerative capacity in cerebral ischemia [15]. The M1 phenotype may exacerbate the post-stroke cerebral damage by releasing pro-inflammatory cytokines [16]. In this research, we found that reduced expression of IL-33 supports neuroprotection by activation of macrophage transformation from M1 to M2, leading to autophagy level downregulation. Over-activated microglia release large amounts of inflammatory cytokines, such as IFN-γ, leading to a severe inflammatory response and exacerbating brain damage [17]. IL-10 is a well-known anti-inflammatory cytokine that has been shown in numerous studies to reduce acute neuronal damage after stroke [18, 19]. Its release has also been proven to exert neuroprotective effects on many immunomodulatory cells, including regulatory T cells and regulatory B cells [20]. This research also showed that decreasing IL-33 suppressed the secretion of pro-inflammatory cytokine (IFN-γ) and stimulated the release of anti- inflammatory cytokines (IL-10 and TGF-β).
Interleukin 33 is an inflammatory mediator that plays a crucial role in the pathogenesis of chronic inflammatory diseases, including bronchial asthma, systemic lupus erythematosus, and other autoimmune diseases [21]. Additionally, a study found that the production of IL-33 in the central nervous system might protect the brain against neuroinflammation by recruiting leukocytes in the brain [22]. Furthermore, in an in vivo experiment, IL-33-deficient mice exhibited reduced lipopolysaccharide (LPS)-induced neuroinflammation [11]. IL-33 also was suggested to be an accelerator of IL-13-dependent autophagy. A study proposed that IL-33 contributed to cerebral protection by mediating the expression of ST2, which is a member of the IL-1 receptor superfamily and widely expressed on various immune cells, including M2 macrophages [23]. However, the relationship between IL-33 and macrophage polarization is still unclear. The current research elucidated how IL-33 regulated the macrophage polarization to trigger I/R injury protection in vitro. When IL-33 was knocked down, the expression of M1-type intracellular markers STAT1, STAT3, IRF5, and IRF8 was significantly downregulated, whereas the expression of M2-type intracellular markers STAT6, PPAR-γ, IRF4, and CEBP-β was up-regulated. Additionally, expression of the autophagy markers Beclin-1 and ULK1 was decreased. Autophagy, also known as type II programmed cell death, is a self-defense mechanism in which lysosomes degrade intracellular metabolic wastes and reuse their degradation products to maintain homeostasis in the cellular environment. Inhibition of overactivated autophagy attenuates cerebral ischemia/reperfusion injury, and improved autophagy ameliorates neuronal apoptosis, whereas autophagy activation leads to permanent cerebral ischemic injury. These results indicate that IL-33 in the OGD/R model affects the relevant signaling pathways by regulating microglia autophagy through macrophage polarization, thereby increasing the degree of injury.
To sum up, the results of this study indicated that IL-33 deficiency alleviates injury and inflammation in the OGD/R model by inhibiting hypoxia-induced autophagy in microglia and promoting microglial polarization toward the anti-inflammatory phenotype (M2). Moreover, IL-33 also regulated hypoxia-induced autophagy in microglia under M1/M2 co-culture conditions. These findings suggest that IL-33 may represent a promising therapeutic target for cerebral I/R injury.