Introduction
Hypereosinophilic syndrome (HES) is a rare and heterogeneous disorder characterized by persistent peripheral blood hypereosinophilia (> 1.5 × 109/l) on at least two separate occasions, accompanied by organ dysfunction or damage that cannot be attributed to other identifiable causes [1]. HES is commonly classified into three categories: neoplastic HES, which is defined by clonal proliferation of eosinophils, often associated with chronic eosinophilic leukemia (CEL) involving the fusion gene FIP1L1+/PDGFRA+; reactive HES, which encompasses various conditions in which eosinophilopoietic factors drive polyclonal eosinophil expansion; and idiopathic HES, in which the underlying mechanism of eosinophilia remains unclear [1].
Lymphocyte variant hypereosinophilic syndrome (L-HES), a subset of reactive HES, is characterized by the presence of immunophenotypically abnormal clonal T cells (most common CD3–CD4+) that produce excessive amounts of eosinophilopoietic cytokines, most notably interleukin 5 (IL-5) [2]. In patients with L-HES, the most common clinical manifestations involve the skin and soft tissues; however, only a few cases of pulmonary infiltrates with eosinophilia have been reported, and even fewer cases present with severe eosinophilic pneumonia (EP) as the sole manifestation of L-HES [3]. Corticosteroids (CS) are recommended as the first-line therapy for L-HES; how- ever, the response rate is lower compared to other subtypes of HES [4]. Among various second-line treatment options, interferon α (IFN- α) appears to be more effective in CS-refractory patients with L-HES [4]. Interleukin 5 (IL-5) plays a central role in the differentiation, activation, and survival of eosinophils [5]; therefore, anti-IL-5 monoclonal antibodies, such as mepolizumab and reslizumab, have been used in the treatment of eosinophilic disorders [6].
Herein, we report a case of a Chinese patient diagnosed with L-HES who presented solely with EP and experienced relapse after receiving long-term CS and IFN- α treatment. However, a promising clinical response was observed after switching to mepolizumab, suggesting that the management of L-HES should be tailored according to the phenotypic characteristics of the disease and the therapeutic efficacy in alleviating symptoms.
Case presentation
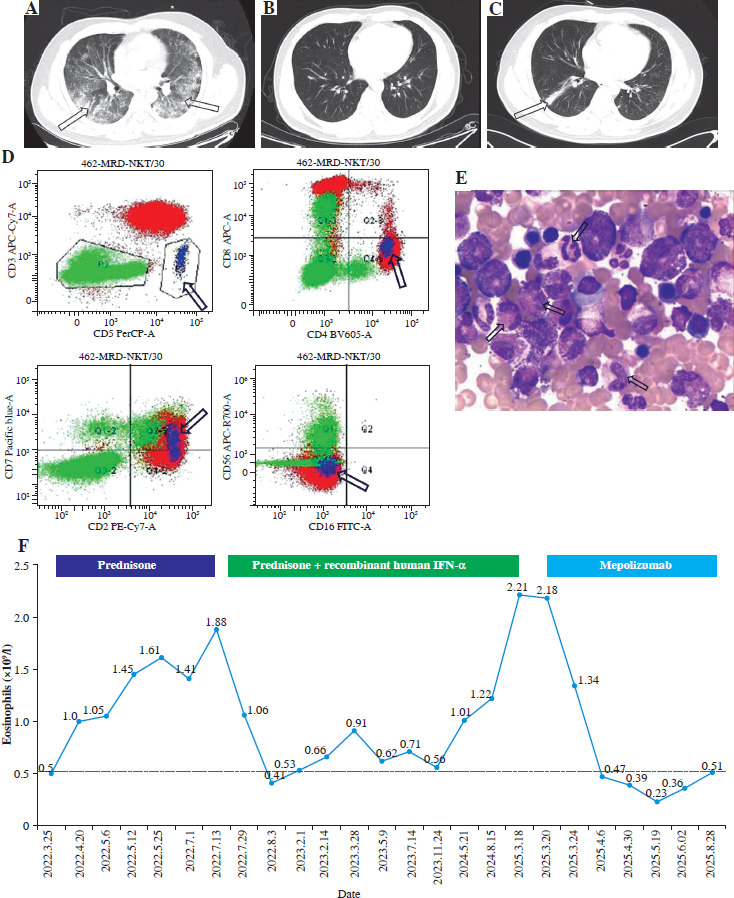
A 35-year-old male patient was referred to our department with a persistent productive cough, dyspnea, and intermittent fever exceeding 38°C for 7 days. The patient did not exhibit pruritus, cutaneous lesions, chest or abdominal pain, hemoptysis, or musculoskeletal pain. Physical examination revealed coarse breath sounds and moist rales in the lower lobes of both lungs. The patient also exhibited hypertension (140/105 mmHg), tachycardia at 126 beats per minute, a peripheral blood oxygen saturation (SpO2) of 92%, and a respiratory rate of 24 breaths per minute. All other physical examination findings were within normal limits. Routine laboratory results are presented in Table 1. Specifically, blood analysis revealed a white blood cell (WBC) count of 22.97 × 109/l and an extremely high eosinophil count of 10.11 × 109/l (44.0% of WBC), with normal levels of hemoglobin and platelets. Arterial blood gas analysis revealed a pH of 7.45, PaO2 of 75.6 mmHg, PaCO2 of 33.5 mmHg, HCO3– of 22.7 mmol/l, and lactate < 1.00 mmol/l. Total IgE was 7630.00 IU/ml. The sputum Gram stain, bacterial cultures, and mycobacterial testing all yielded negative results. Serological tests for etiological agents, including 1,3-β-D-glucan, galactomannan (GM), interferon γ release assay (IGRA), cryptococcus capsular polysaccharide antigen (CRAG), Epstein-Barr virus (EBV) DNA, cytomegalovirus (CMV) DNA, hepatitis B virus (HBV) DNA, and antibodies against HIV, rubella virus, toxoplasma, Schistosoma japonicum, Opisthorchis sinensis, and Echinococcus granulosus, were all negative. Other immunological tests, including anti-neutrophil cytoplasmic antibodies (ANCA, comprising c-ANCA and p-ANCA), IgM, IgA, IgG, IgG4, α-globulin, β-globulin, and γ-globulin, were all within normal limits. Electrocardiography (ECG) demonstrated sinus tachycardia. Chest computed tomography (CT) revealed scattered patchy and streaky opacities, along with some solid densities, involving the interstitial structures of both lungs (Fig. 1A). No significant abnormalities were observed on the abdominal CT scan. Lymphocyte phenotyping by flow cytometry of peripheral blood revealed an aberrant T-cell population accounting for 8% of lymphocytes, which expressed CD2, CD4, CD5, and CD7, but lacked expression of CD3, CD8, and CD16 (Fig. 1D). Bone marrow biopsy and cytomorphology revealed a marked increase in eosinophils (20.5%), with no morphologic dysplasia, increased lymphoma, mastocytosis, or increased plasma cells (Fig. 1E). Biopsy of the left lung revealed that the lung tissue structure was intact, with prominent infiltration of eosinophils and T cells observed in both the alveolar cavity and the interstitial lung, and T-cell receptor (TCR) gene rearrangement was detected. Gene mutations in PDGFRα, PDGFRβ, FGFR1, JAK2, and FLT3 were not detected by next-generation sequencing (NGS) analysis of the patient’s bone marrow sample.
Table 1
Laboratory results in patient with lymphocyte variant hypereosinophilic syndrome (L-HES)
Combining clinical manifestations and laboratory tests, the patient was diagnosed with L-HES and treated with methylprednisolone 40 mg daily for 5 days, which led to marked clinical and laboratory improvement: his dyspnea improved and eosinophil count returned to normal (Fig. 1B, F). He was discharged on prednisone (30 mg daily), but experienced intermittent cough and dyspnea, with eosinophil counts ranging from 1.05 to 1.88 × 109/l (Fig. 1F). Subsequently, recombinant human IFN-α was administered subcutaneously at a dose of 30 µg daily, resulting in partial hematological remission characterized by eosinophil counts ranging from 0.41 to 0.91 × 109/l (Fig. 1F). However, upon slow tapering and discontinuation of IFN-α and prednisone, the patient developed recurrent intermittent cough, and chest CT revealed solid densities in the lower lobe of the right lung (Fig. 1C), accompanied by an increase in eosinophil counts to 2.21 × 109/l (Fig. 1F). The patient subsequently initiated low-dose mepolizumab (100 mg subcutaneously every four weeks) and discontinued IFN-α and prednisone. To date, the patient has achieved complete clinical and hematological remission, with eosinophil counts ranging from 0.23 to 0.51 × 109/l (Fig. 1F).
Fig. 1
A) Chest CT revealed scattered patchy and linear opacities accompanied by areas of solid density (indicated by arrow). B) Following methylprednisolone therapy, follow-up chest CT demonstrated marked regression of pulmonary lesions. C) Upon tapering and discontinuation of IFN-α and prednisone, chest CT showed recurrence of solid densities in the right lower lobe (indicated by arrow). D) Flow cytometric analysis identified an aberrant T-cell population (highlighted in blue) expressing CD2, CD4, CD5, and CD7, but lacking CD3, CD8, and CD16 expression. E) Bone marrow cytomorphology revealed increased eosinophil counts (indicated by arrow). F) Serial monitoring of eosinophil counts was performed throughout the discharge treatment course
Discussion
Eosinophilic pneumonia is a rare disease characterized by marked accumulations of infiltrating eosinophils in the alveolar space and interstitium. It has been reported in myeloid clonal eosinophilia or CEL, reactive eosinophilia (mostly eosinophilic granulomatosis with polyangiitis, EGPA), and idiopathic HES [7]. L-HES is a disorder characterized by the coexistence of clonal and reactive features, in which T lymphocytes with an abnormal immunophenotype produce eosinophilopoietic cytokines, primarily IL-5, leading to reactive eosinophilia. The most frequently observed abnormal immunophenotype of T cells is CD3–CD4+, although CD4+CD7– and CD3+CD4–CD8– TCRαβ+ phenotypes have also been reported [8]. Clinically, patients with L-HES may present with a range of manifestations involving different organs, among which cutaneous findings – such as eczematous dermatitis-like lesions and urticarial plaques – are the most common [4]. To our knowledge, a few other cases of L-HES complicated by pulmonary eosinophilic infiltrates have been described; however, this is the first case in which severe EP is the sole manifestation of L-HES.
There is no established standard therapy for steroid-refractory L-HES. Following disease relapses during treatment with CS and IFN-α, and given the isolated pulmonary involvement, we initiated mepolizumab therapy, which resulted in a favorable clinical response. Mepolizumab has been approved for the treatment of pulmonary eosinophilic disorders such as eosinophilic asthma and EGPA [9]. One study involving 17 patients demonstrated that mepolizumab has a favorable effect on symptoms in L-HES; however, reduced eosinophil-depleting and corticosteroid-sparing effects were observed at the currently approved dose (300 mg every four weeks) [10]. Moreover, while other studies have reported incomplete clinical responses – particularly in skin lesions or dermatitis – even with higher-dose regimens (700 mg or more every four weeks), our patient achieved complete resolution of respiratory symptoms (cough and dyspnea) and a marked reduction in eosinophil counts with low-dose mepolizumab, which was administered safely and well tolerated [11, 12]. This suggests that the therapeutic efficacy of mepolizumab may vary according to the organ systems involved in L-HES; however, a well-designed cohort study is still needed to validate this hypothesis and elucidate the underlying mechanisms. Although mepolizumab appears to have a limited effect on the underlying clonal T cells, given that most L-HES cases are indolent and the risk of lymphoma transformation is relatively low, mepolizumab may offer a more favorable benefit-risk profile compared to lymphodepleting agents such as alemtuzumab [13].
In conclusion, severe EP as the sole manifestation of L-HES is uncommon; however, mepolizumab may represent an effective therapeutic option for this patient population.