Introduction
Bladder cancer is one of the most common malignancies affecting the urinary tract [1]. In 2020, approximately 573 thousand new cases of bladder cancer and 213 thousand related deaths were reported globally [2]. The disease predominantly affects older individuals, with the highest incidence occurring in those over 65 years of age [3]. Bladder cancer is associated with significant morbidity and mortality, often necessitating intensive treatments such as surgery, chemotherapy, and radiation. Despite advancements in these therapeutic strategies, bladder cancer remains a major clinical challenge due to its high recurrence rate and the development of resistance to conventional therapies, contributing to a poor prognosis in advanced stages [4-7].
Dexmedetomidine (Dex) is a selective α2-adrenergic receptor agonist commonly used in clinical practice for sedation and pain relief [8, 9]. Recent studies have shown that Dex possesses potential antitumor effects, including in esophageal cancer and rectal cancer [10, 11]. Ferroptosis, an iron-dependent form of regulated cell death distinct from apoptosis and necrosis, is important in cancer progression and therapy resistance [12, 13]. The Wnt/β-catenin signaling pathway is known to be hyperactivated in various cancers, including bladder cancer. Dysregulation of this pathway is associated with tumor growth, immune evasion, and resistance to cell death [14]. A study found that Dex could modulate ferroptosis in animal models [15]. Additionally, Dex suppressed ovarian cancer progression by inhibiting the Wnt/β-catenin signaling pathway, leading to reduced cell proliferation, invasion, and migration, as well as increased apoptosis [16].
Based on this background, we hypothesized that Dex exerts its antitumor effects by inhibiting the Wnt/β-catenin pathway, regulating ferroptosis in bladder cancer cells, and modulating the tumor immune microenvironment. The aim of this study was to explore the role of Dex in bladder cancer progression, focusing on its potential to induce ferroptosis, alter immune responses, and suppress tumor growth in a xenograft mouse model.
Material and methods
Cell culture and treatment
Peripheral blood mononuclear cells (PBMCs), SV-HU-1, T24, and RT4 cells (all purchased from ATCC) were cultured under standard conditions. PBMCs were cultured in RPMI-1640 medium, whereas SV-HU-1, T24, and RT4 cells were grown in DMEM with 10% fetal bovine serum, 50 U/ml penicillin, and 50 µg/ml streptomycin at 37°C with 5% CO2. The medium was refreshed two to three times per week. T24 and RT4 bladder cancer cells were plated at 1.5 × 105 cells per well in 24-well plates and treated with different concentrations of Dex (0 to 4 µM, Sigma-Aldrich, USA) for 24 hours to investigate its effect on cell proliferation, ferroptosis, and immune response modulation.
In specific experimental groups, additional treatments were applied as follows: 1) Dex + erastin (Selleckchem, USA): Cells were treated with 2 µM Dex combined with 5 µM erastin to enhance ferroptotic cell death; 2) Dex + ferrostatin-1 (Sigma-Aldrich, USA): Cells were treated with 2 µM Dex combined with 5 µM ferrostatin-1 to assess the reversal of ferroptosis; 3) Dex + LiCl (Sigma-Aldrich, USA): Cells were treated with 2 µM Dex in combination with 20 mM LiCl [17] to evaluate the role of the Wnt/β-catenin pathway in Dex-mediated effects. The concentration of 2 µM was selected based on a study that used similar concentrations to investigate the effects of Dex on cancer cells [18]. This concentration was effective in inducing ferroptosis and modulating key signaling pathways, such as the Wnt/β-catenin pathway, without significantly affecting cell viability in bladder cancer cell lines. LiCl is a commonly used activator of the Wnt/β-catenin signaling pathway in cell biology. It functions by inhibiting glycogen synthase kinase-3β (GSK-3β), leading to the accumulation of β-catenin and the subsequent activation of the Wnt signaling pathway. For immune modulation studies, T24 and RT4 cells were co-cultured with PBMCs at a 1 : 10 ratio (cancer cells to PBMCs) for 24 hours following Dex (2 µM) treatment.
Cell Counting Kit-8 (CCK-8)
The cells were plated in 96-well plates at a density of 5 × 103 cells/well. After incubating the cells with CCK-8 reagent (10 µl, Sangon) for 2 hours, the absorbance was measured at 450 nm using a microplate spectrophotometer to determine cell proliferation.
Measurement of Fe2+
Intracellular Fe2+ was measured using a commercial kit and FerroOrange (DojinDo, Japan) as per the manufacturer’s instructions. Cells were seeded in confocal dishes, treated with 1 µmol/l FerroOrange in PBS at 37oC for 30 minutes, and then observed under a confocal laser scanning microscope (Olympus FV3000). The average fluorescence intensity was analyzed using ImageJ software.
Measurement of lipid ROS
Lipid ROS levels were assessed using the BODIPY 581/591 C11 probe (Thermo Fisher Scientific). Cells (1 × 104 per group) were seeded in confocal dishes a day before the experiment and incubated with 5 µM BODIPY 581/591 C11 and Hoechst in PBS at 37oC for 15 minutes. Confocal imaging was performed with an Olympus FV3000 microscope, and the ratio of oxidized (green) to total fluorescence (green + red) was calculated.
Immunofluorescence (IFC)
T24 and RT4 cells were treated with Dex (0, 0.5, 1, and 2 µM) (Sigma-Aldrich) for 24 hours. After treatment, cells were incubated with 10 µM DCFH-DA (Beyotime) for 30 minutes to stain for ROS. After washing with PBS, cells were fixed with 4% paraformaldehyde (Beyotime) for 15 minutes and counterstained with 1 µg/ml DAPI (Beyotime) for 10 minutes. Fluorescence images were captured using a Leica fluorescence microscope. Fluorescence intensity was quantified using ImageJ software. At least five random fields per group were analyzed, and the mean fluorescence intensity (MFI) was calculated and normalized to the control (0 µM Dex).
Flow cytometry analysis
Cells were collected and stained with PE-conjugated anti-CD8 antibody (BD Biosciences) for 30 minutes at 4oC in the dark. After staining, they were washed and resuspended in 500 µl of PBS. Data were collected and analyzed with FlowJo software to determine the percentage of CD8+ T cells in the co-culture system.
ELISA
Samples were centrifuged at 10,000 × g for 1 minute, the supernatant was discarded, and the wells were washed with 0.1% PBST. Blocking was performed with 1% BSA in PBS, and the plate was incubated overnight at 4°C. After discarding the supernatant, the wells were washed again with 0.1% PBST. Serum samples were diluted 1 : 50 in blocking solution, with PBS used as a negative control. The samples were incubated at 37°C for 1 hour and washed three times with PBS. Then, 100 µl of freshly diluted enzyme-labeled antibody was added to each well, and the plate was incubated at 37°C for 1 hour. After washing the wells three times with PBS, 100 µl of TMB substrate solution was added and incubated at 37°C for 30 minutes. The reaction was terminated by adding 50 µl of 2 mM sulfuric acid. The optical density (OD) was measured at 450 nm using a SpectraMax iD5 microplate reader.
Tumorigenicity assay
BALB/c nude mice (4 weeks old, 13-15 g) were housed under pathogen-free conditions (20-26°C, 40-70% humidity, 12-hour light/dark cycle) with access to food and water. The xenograft tumor model was established according to the method of Dewangan et al. Cultured T24 cells (1 × 107) were subcutaneously injected into the mice. The mice were then randomly divided into two groups: the control group and 2.0 µg/kg Dex group (Dex was administered intraperitoneally at 2.0 µg/kg, once a day for 15 days), with 6 mice in each group. The 2.0 µg/kg concentration used for the in vivo study was based on our previous optimization experiments with Dex to ensure that it was within the therapeutic range for the animal model. This dose was consistent with the results of a previous animal study involving Dex [18], in which Dex was shown to have antitumor effects. Tumor volume was determined by measuring the length (l) and width (w) of the tumors and calculated using the formula: V = lw2/2. After 35 days, the mice were euthanized by CO2 inhalation, and the tumors were excised for photographing and weighing. The experimental protocol was approved by the Animal Ethics Committee of our institute, and all procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Immunohistochemistry (IHC)
Tumor tissues were fixed in 4% paraformaldehyde, dehydrated, paraffin-embedded, and sliced into 4-µm sections. After deparaffinization, they were incubated overnight at 4°C with primary antibodies, then with secondary antibodies for 30 minutes at 37°C. HRP-conjugated solution and DAB staining were then applied for 5-10 minutes. After hematoxylin counterstaining, the tissues were mounted, dried, and photographed. Five high-power fields were selected for observation and quantification.
Western blot (WB)
Total proteins were extracted from both the cells and tissue sections. The Pierce BCA Protein Assay Kit (Thermo Scientific) was used for protein quantification. Subsequently, 20 µg of the extracted proteins were separated using SDS-PAGE and transferred to PVDF membranes. Following overnight incubation with a primary antibody at 4°C and subsequent washing, the membranes were incubated with Goat Anti-Rabbit IgG H&L secondary antibody (ab96899, 1/1000, Abcam) at 37°C for 45 minutes. The primary antibodies used were: GPX4 antibody (ab125066, 1/1000, Abcam), SLC7A11 antibody (ab307601, 1/1000, Abcam), PD-L1 antibody ab228415, 1/1000, Abcam), active β-catenin antibody (ab305261, 1/1000, Abcam), β-catenin antibody (ab32572, 1/5000, Abcam), c-Myc antibody (ab185656, 1/2000, Abcam), cyclin D1 antibody (ab134175, 1/10000, Abcam), GAPDH antibody (ab8245, 1/1000, Mybiosource), and GAPDH antibody (Abcam, USA). To visualize the protein bands, we used the Pierce ECL Western Blotting Substrate, which was procured from Pierce in Shanghai, China. The membranes were developed and imaged using a chemiluminescence imaging system.
Results
Dexmedetomidine inhibited bladder cancer cell proliferation
The effect of Dex on cell viability was assessed in normal bladder cells (SV-HU-1) and bladder cancer cell lines (T24 and RT4) using a CCK8 assay after 24 hours of treatment with different concentrations of Dex. In SV-HU-1 cells, cell viability showed no significant decrease at concentrations ≤ 2 µM, indicating that Dex is non-toxic to normal bladder cells within this dosage range (Fig. 1A).
Fig. 1
Dexmedetomidine inhibited the proliferation of bladder cancer cells. A) SV-HU-1 normal bladder cells, B) T24 bladder cancer cells, and C) RT4 bladder cancer cells were treated with increasing concentrations of dexmedetomidine (0, 0.125, 0.25, 0.5, 1, 2, 4 μM) for 24 hours, and cell viability was measured using the CCK8 assay. Data are presented as mean ± SD from three independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s posthoc test. *p < 0.05, **p < 0.01 compared to the control group (0 μM Dex)
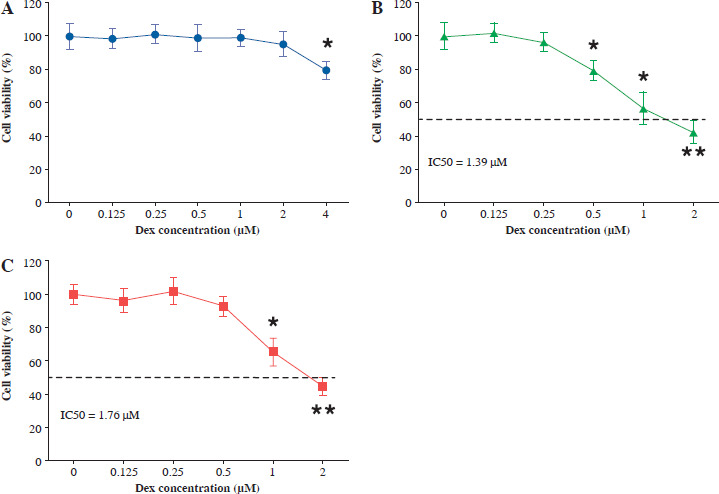
In T24 bladder cancer cells, a significant reduction in cell viability was observed at a Dex concentration of 0.5 µM (Fig. 1B). In contrast, RT4 bladder cancer cells showed no significant decrease in cell viability at 0.5 µM, with a marked reduction only observed at 1 µM and above (Fig. 1C). These results suggested that Dex selectively inhibited the proliferation of bladder cancer cells (T24 and RT4), while exerting minimal effects on normal bladder cells (SV-HU-1).
Dexmedetomidine induced ferroptosis and increased ROS levels
Dexmedetomidine significantly reduced the expression of SLC7A11 and GPX4 in T24 and RT4 cells, with the most pronounced reduction observed at the higher concentrations of 1 µM and 2 µM (Fig. 2A). Similarly, Dex increased intracellular Fe2+ levels in both cell lines, with the greatest increase seen at 1 µM and 2 µM (Fig. 2B). The addition of erastin further elevated Fe2+ levels, while ferrostatin-1 reversed the Dex-induced increase (Fig. 2C). Dex treatment also led to a marked increase in lipid ROS levels in T24 and RT4 cells, particularly at 1 µM and 2 µM concentrations, as demonstrated by the quantitative analysis (Fig. 2D). Immunofluorescence staining revealed a clear rise in ROS accumulation in T24 and RT4 cells, as indicated by the stronger green fluorescence at 1 µM and 2 µM Dex (Fig. 2E). These results indicated that Dex promoted ferroptosis in bladder cancer cells by increasing intracellular iron levels and ROS production.
Fig. 2
Dexmedetomidine induced ferroptosis and increased ROS levels in bladder cancer cells. A) Western blot analysis of GPX4 and SLC7A11 expression in T24 and RT4 cells treated with different concentrations of Dex (0, 0.5, 1, 2 μM) for 24 hours. B) Relative Fe2+ levels in T24 and RT4 cells treated with varying Dex concentrations. C) Fe2+ levels in RT4 and T24 cells treated with Dex (2 μM) alone or in combination with erastin (ferroptosis inducer) or ferrostatin-1 (ferroptosis inhibitor). D) Lipid ROS levels in T24 and RT4 cells after treatment with increasing Dex concentrations. Data are presented as mean ± SD from three independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. *p < 0.05, **p < 0.01 compared to the control group (0 μM Dex) E) Immunofluorescence images showing ROS accumulation in T24 and RT4 cells treated with different Dex concentrations
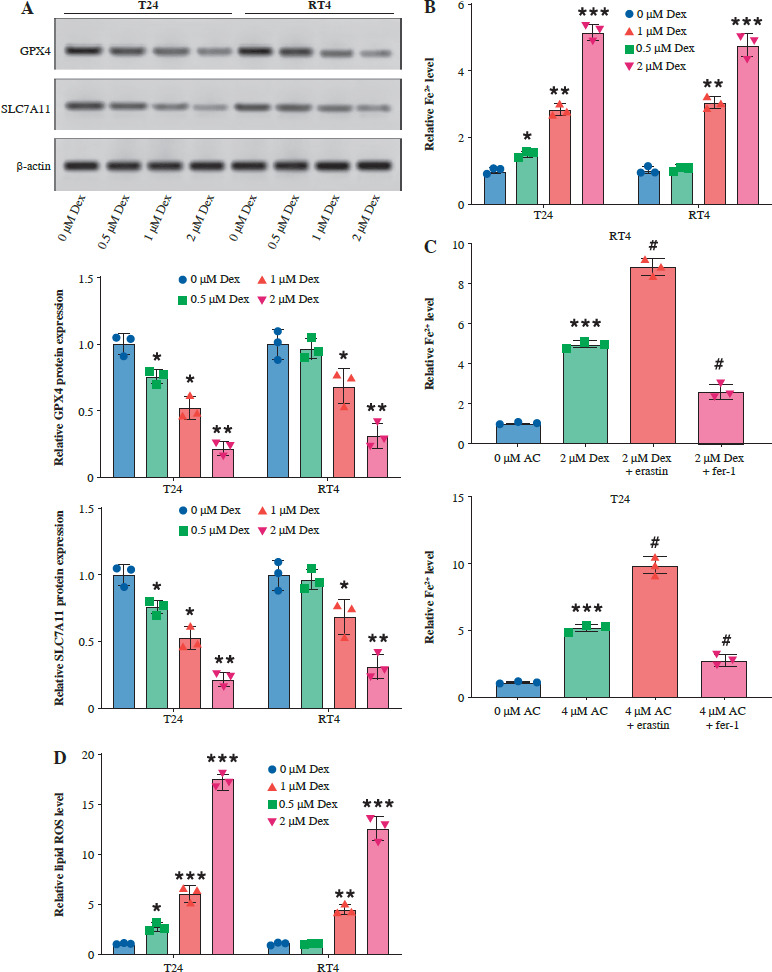
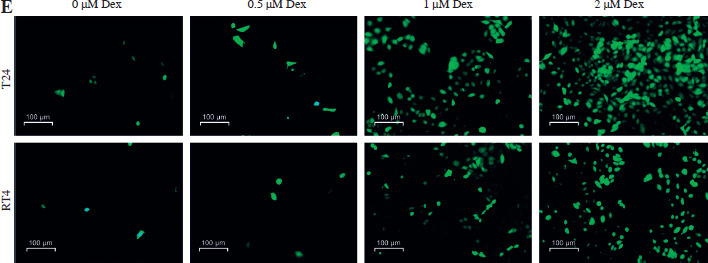
Dexmedetomidine inhibited immune evasion
Subsequently, we examined the effect of Dex on PD-L1 expression in bladder cancer cells and CD8+ T-cell activity to evaluate its potential in inhibiting tumor immune evasion. Western blot analysis showed that Dex treatment significantly reduced PD-L1 protein expression in both T24 and RT4 cells, with more pronounced reductions observed at 1 µM and 2 µM concentrations (Fig. 3A). Additionally, Dex treatment increased the percentage of CD8+ T cells co-cultured with T24 and RT4 cells (Fig. 3B, C). Furthermore, ELISA results showed that Dex treatment significantly reduced interleukin (IL)-10 levels and increased interferon γ (IFN-γ) levels (Fig. 3D) in the co-cultures of PBMCs with T24 and RT4 cells. These findings suggested that Dex inhibited immune evasion by reducing PD-L1 expression and enhancing CD8+ T-cell activation in bladder cancer cells.
Fig. 3
Dexmedetomidine reduced PD-L1 expression and enhanced CD8+ T-cell activity in bladder cancer cells. A) Western blot analysis of PD-L1 expression in T24 and RT4 cells treated with different concentrations of Dex (0, 0.5, 1, 2 μM) for 24 hours. B) Representative flow cytometry plot showing CD3+CD8+ T-cell percentages in co-cultures of PBMCs with T24 or RT4 cells treated with 0 μM or 2 μM Dex. C) Quantification of CD8+ T-cell percentages in PBMC co-cultures with T24 and RT4 cells. D) IFN-γ and IL-10 levels in PBMC co-cultures with T24 and RT4 cells treated with 0 μM or 2 μM Dex, measured by ELISA. Data are presented as mean ± SD from three independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. *p < 0.05, **p < 0.01 compared to the control group (0 μM Dex)
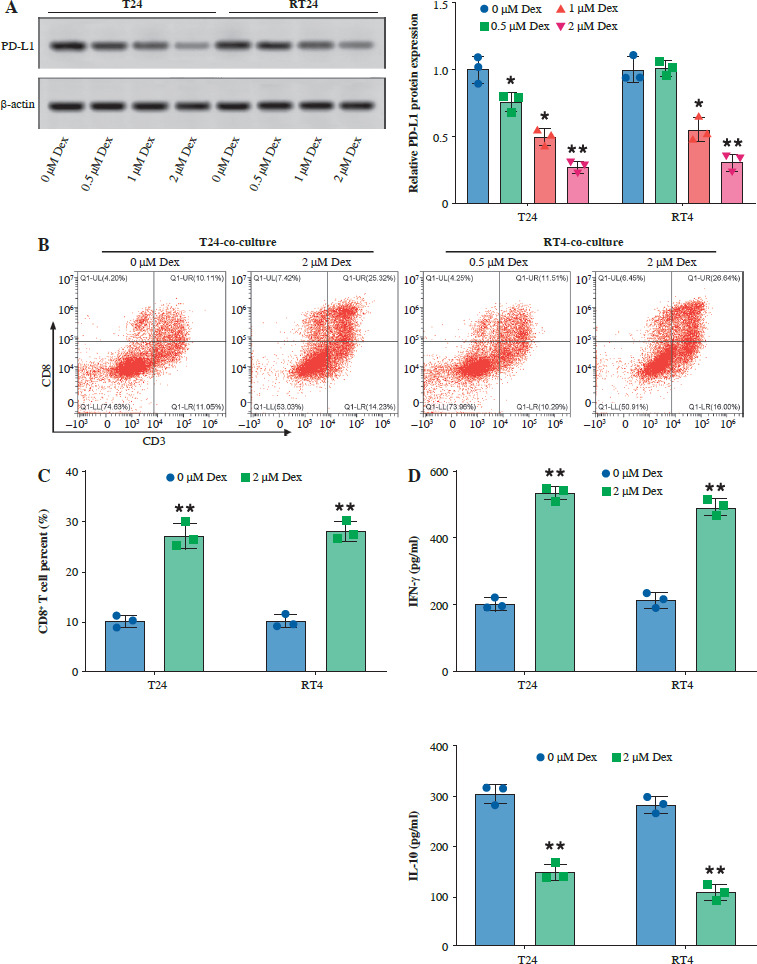
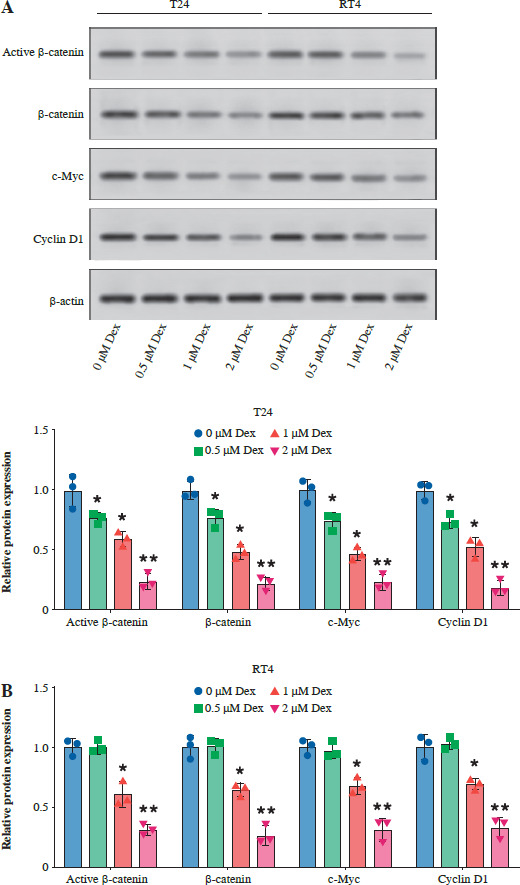
Dexmedetomidine inhibited Wnt/β-catenin signaling
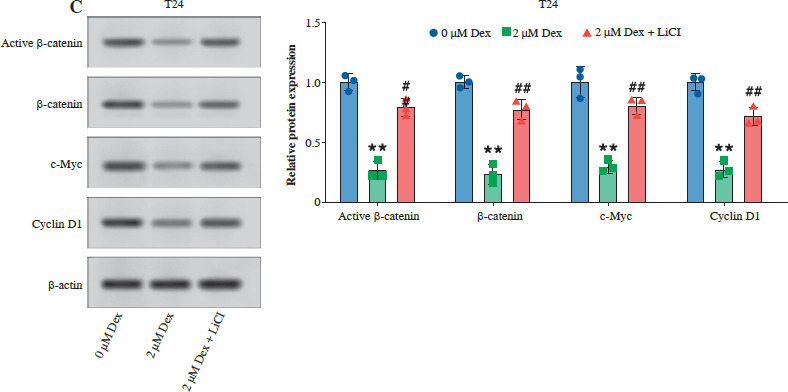
Then, we examined the effect of Dex on the Wnt/β-catenin pathway by analyzing the expression of active β-catenin, β-catenin, c-Myc, and cyclin D1 in T24 and RT4 bladder cancer cells. As shown in Figure 4A, B, Dex treatment significantly reduced the levels of active β-catenin, c-Myc, and cyclin D1 in both T24 and RT4 cells, with more pronounced reductions observed at 1 µM and 2 µM. In a further experiment, the results of WB showed that the combination with LiCl partially reversed the Dex-induced reduction in active β-catenin, c-Myc, and cyclin D1 expression, while total β-catenin expression remained unchanged (Fig. 4C). These results indicated that Dex suppressed Wnt/β-catenin signaling in bladder cancer cells.
Fig. 4
Dexmedetomidine inhibited Wnt/β-catenin signaling in bladder cancer cells. A) Western blot analysis of active β-catenin, β-catenin, c-Myc, and cyclin D1 expression in T24 and RT4 cells treated with different concentrations of Dex (0, 0.5, 1, 2 μM) for 24 hours. B) Western blot analysis of active β-catenin, β-catenin, c-Myc, and cyclin D1 expression in RT4 cells. Data are presented as mean ± SD from three independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. *p < 0.05, **p < 0.01 compared to the control (0 μM Dex); #p < 0.05, ##p < 0.01 compared to the 2 μM Dex group C) Western blot analysis and quantification of active β-catenin, β-catenin, c-Myc, and cyclin D1 in T24 cells treated with 2 μM Dex alone or in combination with 2 μM LiCl (Wnt/β-catenin activator). Data are presented as mean ± SD from three independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. *p < 0.05, **p < 0.01 compared to the control (0 μM Dex); #p < 0.05, ##p < 0.01 compared to the 2 μM Dex group
Dexmedetomidine regulated ferroptosis and immune evasion through inhibition of the Wnt/β-catenin pathway
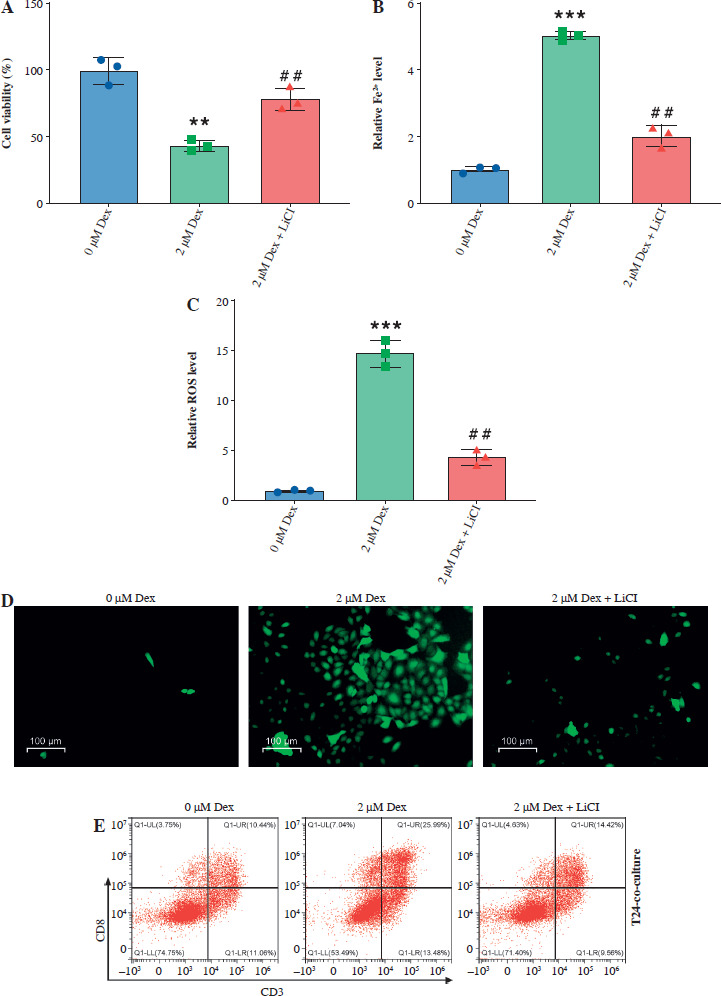
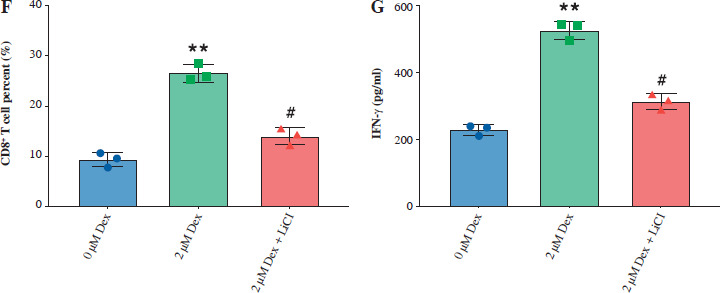
Then, we assessed the role of Dex on bladder cancer cell proliferation, ferroptosis, and immune response modulation, both alone and in combination with the Wnt/β-catenin activator LiCl. Dex treatment significantly reduced cell viability in T24 cells, while the addition of LiCl partially reversed this reduction (Fig. 5A). Similarly, Dex elevated intracellular Fe2+ levels and ROS accumulation, and LiCl attenuated these effects (Fig. 5B, C). Immunofluorescence images confirmed the increase in ROS levels with Dex treatment and the partial reduction by LiCl (Fig. 5D). Dex treatment increased the percentage of CD8+ T cells in co-culture with PBMCs, while LiCl addition reduced this increase (Fig. 5E, F). Finally, the ELISA results indicated that Dex significantly elevated IFN-γ levels in the co-culture supernatants, and this elevation was partially reversed by LiCl (Fig. 5G). These findings suggested that Dex promoted ferroptosis and enhanced immune responses in bladder cancer cells, with the Wnt/β-catenin pathway playing a crucial regulatory role.
Fig. 5
Dexmedetomidine regulated ferroptosis and immune evasion in bladder cancer cells through inhibition of the Wnt/β-catenin pathway. A) CCK-8 assay measuring cell viability in T24 cells. B) Kit-based detection of Fe2+ levels in T24 cells. C) ROS levels in T24 cells. D) Immunofluorescence images of ROS levels in T24 cells treated with Dex and Dex + LiCl. E) Flow cytometry analysis showing CD8+ T-cell percentages in PBMC co-cultures with T24 cells treated with Dex and Dex + LiCl. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. **p < 0.01 compared to control (0 μM Dex); #p < 0.05, ##p < 0.01 compared to the 2 μM Dex group F) Quantification of CD8+ T-cell percentages. G) IFN-γ levels measured by ELISA in PBMC-T24 cocultures. Data are presented as mean ± SD from three independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. **p < 0.01 compared to control (0 μM Dex); #p < 0.05, ##p < 0.01 compared to the 2 μM Dex group
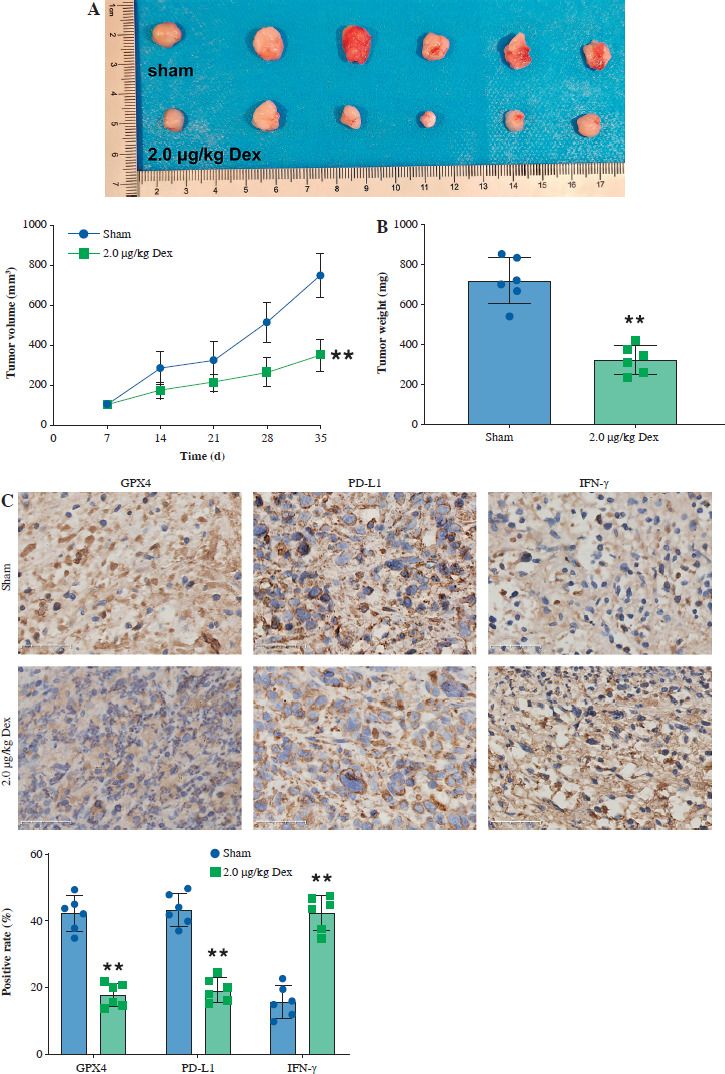
Dexmedetomidine inhibited tumor growth in vivo
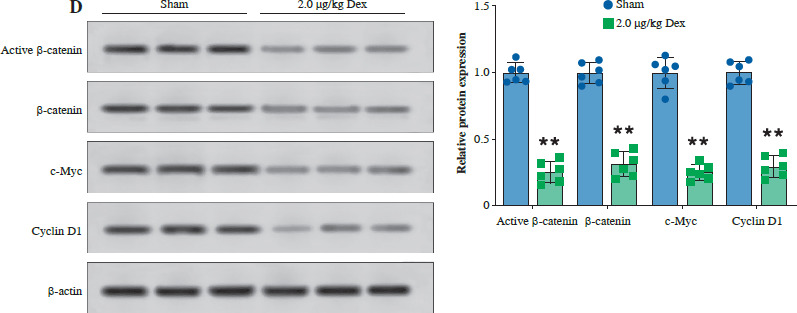
Finally, we evaluated the effect of Dex on bladder cancer tumor growth in a T24 subcutaneous xenograft mouse model. Tumor volume was significantly reduced in mice treated with 2.0 µg/kg Dex compared to the sham group (Fig. 6A). Additionally, tumor weight was significantly lower in the Dex-treated group (Fig. 6B). Immunohistochemical analysis revealed that Dex treatment reduced the expression of GPX4 and PD-L1, while increasing IFN-γ levels in tumor tissues (Fig. 6C). Western blot analysis showed that Dex reduced the expression of active β-catenin, c-Myc, and cyclin D1 in tumor tissues, while the levels of total β-catenin remained unchanged (Fig. 6D). These findings suggest that Dex inhibits tumor growth in vivo by promoting ferroptosis and modulating the immune response, potentially through inhibition of the Wnt/β-catenin signaling pathway.
Fig. 6
Dexmedetomidine inhibited tumor growth in vivo. A) Tumor images and tumor volume growth curves in mice. B) Tumor weight measured at the end of the experiment. C) Immunohistochemical analysis of GPX4, PD-L1, and IFN-γ expression in tumor tissues. Data are presented as mean ± SD from five mice per group. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. **p < 0.01 compared to sham D) Western blot analysis of active β-catenin, β-catenin, c-Myc, and cyclin D1 expression in tumor tissues. Data are presented as mean ± SD from five mice per group. Statistical significance was determined using one-way ANOVA followed by Tukey’s post-hoc test. **p < 0.01 compared to sham
Discussion
Bladder cancer is associated with significant morbidity and mortality, often necessitating intensive treatments such as surgery, chemotherapy, and radiation, yet it remains a major clinical challenge due to its high recurrence rate and resistance to conventional therapies [19]. This study demonstrated that Dex exerted significant inhibitory effects on bladder cancer progression, primarily through the induction of ferroptosis and modulation of the Wnt/β-catenin pathway.
Dexmedetomidine, widely known for its sedative and analgesic properties, has recently been explored for its potential antitumor effects. A study on an ovarian cancer mouse model showed that Dex reduced tumor burden by improving postoperative natural killer (NK) cell activity and lowering cortisol and tumor necrosis factor α (TNF-α) levels, which may suppress the surgical stress response and its associated tumor growth [20]. Additionally, in a lung cancer xenograft model, Dex was found to inhibit cancer progression by enhancing NK cell activity, reducing proinflammatory cytokines, and suppressing inflammasome-related signaling, further supporting its potential role in cancer treatment [21]. Recently, its role in regulating ferroptosis has also been explored. Gao et al. found that DEX mediates ferroptosis by regulating the circ0008035/miR-302a/E2F7 axis [18]. In this study, we similarly found that Dex can induce ferroptosis in bladder cancer cells by increasing intracellular iron (Fe2+) levels and enhancing ROS, both hallmark signals of ferroptotic cell death. Furthermore, our data showed that Dex treatment significantly reduced expression of SLC7A11 and GPX4, two key regulators of the ferroptotic response. The inhibition of SLC7A11 and GPX4 by Dex disrupts the antioxidant defenses of the cells, making bladder cancer cells more susceptible to oxidative stress and ferroptotic cell death [22]. This mechanism suggests that Dex not only directly induces ferroptosis but also alters the oxidative balance within cancer cells, pushing them toward cell death.
Notably, ferroptosis has been increasingly recognized as a modulator of the tumor immune microenvironment. As a form of regulated cell death, ferroptosis can lead to the release of damage-associated molecular patterns (DAMPs), which activate innate immune responses, including the recruitment and activation of dendritic cells and CD8+ T cells [23, 24]. This immune activation enhances the recognition and clearance of tumor cells [25, 26]. In our study, we observed that Dex not only promoted ferroptosis but also modulated the immune microenvironment by reducing PD-L1 expression and enhancing CD8+ T-cell activity. This suggests that ferroptosis induced by Dex may contribute to immune system activation, enhancing its antitumor effects by increasing immune cell infiltration and promoting an immune response against the tumor. Therefore, the effects of Dex on bladder cancer progression appear to involve both ferroptosis and immune modulation, working synergistically to suppress tumor growth. The combination of ferroptotic cell death and immune activation offers a potential therapeutic strategy for enhancing the antitumor response in bladder cancer.
The Wnt/β-catenin pathway is a well-known regulator of cell proliferation, differentiation, and survival [27]. For instance, Jiménez-Guerrero et al. demonstrated that hyperactivation of Wnt/β-catenin contributed to chemotherapy resistance and the maintenance of cancer stem cell-like properties in bladder cancer [28]. Additionally, another study revealed that Wnt/β-catenin activation promoted bladder cancer metastasis through the regulation of epithelial–mesenchymal transition (EMT), further underscoring the pathway’s role in cancer progression [29]. In this study, we found that Dex effectively inhibited the Wnt/β-catenin pathway. This downregulation of Wnt/β-catenin signaling likely contributed to the suppression of cancer cell proliferation observed in our experiments. By inhibiting this pathway, Dex disrupts the oncogenic signaling necessary for bladder cancer cell survival and growth. Importantly, the link between Wnt/β-catenin signaling and ferroptosis is an area of active research. Suppression of Wnt/β-catenin exacerbates ferroptosis in melanoma by promoting lipid peroxidation [29]. Similarly, inhibition of the Wnt/β-catenin pathway was found to induce ferroptosis by downregulating key ferroptosis-related proteins, highlighting the role of Wnt/β-catenin signaling in ferroptosis regulation [30]. In our study, Dex’s inhibition of the Wnt/β-catenin pathway correlated with increased markers of ferroptosis, suggesting a dual mechanism where Dex not only inhibits cell proliferation via Wnt/β-catenin suppression but also sensitizes cells to ferroptotic death. This dual action makes Dex a particularly potent agent in targeting bladder cancer, as it can simultaneously inhibit tumor growth and promote ferroptotic cell death.
In addition to its direct effects on ferroptosis and the immune microenvironment, Dex may have synergistic effects when combined with other established bladder cancer treatments. Notably, the combination of Dex with PD-L1 inhibitors could enhance immune activation by both promoting ferroptosis and blocking immune checkpoint pathways. Previous studies have shown that blocking PD-L1 can enhance CD8+ T-cell activity and increase tumor cell recognition [26, 31]. Dex’s ability to downregulate PD-L1 expression and promote ferroptosis could further amplify this effect, potentially leading to a more robust antitumor immune response. This synergy could provide a promising therapeutic strategy in bladder cancer treatment. Furthermore, the Wnt/β-catenin signaling pathway, which is disrupted by Dex, plays a key role in regulating cell survival, proliferation, and migration in bladder cancer. Future studies could explore whether Dex’s inhibition of Wnt/β-catenin signaling also enhances immune responses in the tumor microenvironment by modulating immune cell infiltration and cytokine production. While our study focused on the relationship between Dex and ferroptosis, we hypothesize that Dex’s effects on the Wnt/β-catenin pathway might contribute to its antitumor efficacy through immune regulation. The mechanistic interplay between Dex, ferroptosis, and Wnt/β-catenin signaling warrants further investigation.
Despite the promising findings, this study has several limitations that warrant further exploration. First, the in vitro nature of the majority of our experiments may not fully capture the complexity of tumor-immune interactions in a living organism. While the mouse xenograft model provided valuable insights into the in vivo effects of Dex, future studies using genetically engineered models or patient-derived xenografts could offer a more accurate representation of Dex’s antitumor efficacy in human bladder cancer. Second, although we observed significant inhibition of the Wnt/β-catenin pathway and induction of ferroptosis, the precise molecular interactions between these two processes remain unclear. Further mechanistic studies are needed to elucidate how Dex simultaneously targets both pathways and whether there are specific upstream regulators that mediate this effect. Additionally, investigating whether Dex-induced ferroptosis is dependent on the inhibition of Wnt/β-catenin signaling, or if these are parallel but independent mechanisms, would provide more clarity.
Conclusions
In conclusion, this study highlighted the potential of Dex as a novel therapeutic agent for bladder cancer. By targeting both the Wnt/β-catenin signaling pathway and ferroptosis, Dex not only inhibited cancer cell proliferation but also promoted cancer cell death through oxidative stress. Additionally, its modulation of the tumor immune microenvironment further enhanced its antitumor effects, suggesting that Dex could be a valuable adjunct in cancer immunotherapy. However, further research is needed to explore the synergistic effects of Dex with other established bladder cancer treatments, particularly immune checkpoint inhibitors such as PD-L1 inhibitors. Additionally, investigating the role of Dex and Wnt/β-catenin in various bladder cancer subtypes could provide valuable insights into its broader therapeutic potential. Future studies should focus on delineating the mechanistic connections between ferroptosis, immune modulation, and the Wnt/β-catenin signaling pathway to optimize Dex-based therapies for bladder cancer treatment.