
Introduction
Immune checkpoint inhibitors (ICIs) targeting programmed cell death protein 1 (PD-1) or its ligand 1 (PD-L1) have a wide range of applications in the treatment of tumors [1, 2]. Although PD-1/PD-L1 significantly improves the prognosis of oncology patients, it brings with it a variety of immune-related adverse events (irAE) that are gaining increasing attention, the most fatal of which is cardiotoxic reactions [3, 4]. Cardiotoxic reactions include a variety of diseases, such as myocarditis, cardiomyopathy and cardiac fibrosis. Among them, myocarditis combined with cardiomyocyte apoptosis is one of the most important clinical and pathological features [5-7]. The pathological mechanisms of irAE include multiple factors, such as direct cytotoxic activity of T cells against non-tumor cells and the release of large amounts of inflammatory factors. Therefore, reducing systemic inflammation overall leads to a significant improvement in ameliorating PD-1/PD-L1 inhibitor-induced cardiotoxicity.
There are a number of immunotherapy drugs, including nivolumab and pembrolizumab, that have been approved by the U.S. Food and Drug Administration for cancer treatment [8]. BMS-1, a small molecule inhibitor of PD-1/PD-L1-related effects, has similar effects to anti-PD-1/PD-L1 antibodies, showing that it can replace the antibodies used in immunotherapy [9-11]. In addition, compared with antibody drugs, small molecule PD-1/PD-L1 inhibitors offer valuable pharmacological advantages and can be used as PD-1/PD-L1 inhibitors in basic research [12, 13].
The suppressor of cytokine signaling 3 (SOCS3) was found to be responsible for cell motility via regulation of Janus kinase/signal transducer and activator of transcription 3/interleukin 6 (JAK/STAT/IL-6) cascades [14]. It is well documented that granulocyte colony stimulating factor activation (G-CSF) by its binding in myeloid cells leads to activation of downstream signaling cascades, including the Janus kinase, signal transducer and activator of transcription 3, suppressor of cytokine signaling 3 (JAK/STAT/SOCS) pathway [15]. It induces inflammatory tolerance in the body and suppresses neuroinflammation by inducing SOCS3 expression [16]. SOCS3 is an important negative regulator of various cytokine signaling pathways, such as interleukin (IL)-1b, tumor necrosis factor α (TNF-α) and IL-6 signaling pathways [17-20]. Therefore, in our study we further investigated the role of SOCS using PD-1/PD-L1 inhibitors.
In addition, macrophages are the main producers of inflammatory mediators in autoimmune diseases and auto- inflammatory diseases [21]. Macrophage dysfunction can lead to abnormal repair, allowing uncontrolled production of inflammatory mediators and growth factors. Inadequate production of anti-inflammatory macrophages, or failure of communication between macrophages and epithelial cells, endothelial cells, fibroblasts, and stem or tissue progenitor cells, contributes to a state of ongoing injury, which may lead to the development of pathological fibrosis [22, 23].
In the present study, we first analyzed the cardiotoxic effects produced by PD-1/PD-L1 inhibitors using melanoma mice. Then we investigated whether macrophage- derived factor IL-1b and TNF-α contribute to the apoptotic and cytotoxic effects induced by PD-1/PD-L1 inhibitors in cardiomyocytes. Finally, we analyzed the role of the SOCS3/JAK/STAT3 signaling pathway among others. Our study contributes to elucidating the novel mechanism of action of PD-1/PD-L1 inhibitor-associated cardiotoxicity and may provide potential therapeutic targets.
Material and methods
This study was approved by the ethics committee of the Affiliated Cancer Hospital and Institute of Guangzhou Medical University and the institutional review board (IRB) (GZZL2015.5115.25). All animal experiments complied with the ARRIVE guidelines and were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments, or the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Cell culture
The National Cheng Kung University (NCKU) Hospital’s Department of Internal Medicine generously donated the Rockefeller University embryonic stem cell line-2 (RUES2). Growth factor-reduced Matrigel (#354230; Corning, Carlsbad, Leicestershire, UK) was applied to the culture plate as an extracellular matrix covering before the cells were cultured. Then, cells were plated on Matrigel-coated plates that also included essential-8 (E8) media and 5 M Y27632 (#A1517001; Life Technologies, Carlsbad, CA, USA) (a ROCK inhibitor 1254/1; R&D Systems, Minneapolis, Minnesota, USA). Daily E8 medium renewal without Y27632 was carried out. Every two to three days, RUES2 was passaged utilizing Innovative Cell Technologies’ Accutase (San Diego, CA, USA) for cell detachment.
The differentiation of cardiomyocytes was carried out as previously reported [24]. RUES2 cells were briefly multiplied to create a monolayer on the plate. The next day, the medium was supplemented with E8 medium and 1 M CHIR99021 (a GSK-3 inhibitor; Tocris Bioscience, Bristol, UK). The first day of distinction was at this time. On day 0, the medium was changed to RPMI 1640 medium supplemented with 2% of B-27 without insulin (Invitrogen, Carlsbad, CA) and 100 ng/ml activin A (R&D Systems). The medium was changed to RPMI/B-27 without insulin supplemented with 5 ng/ml bone morphogenic protein-4 and 1 M CHIR99021 after 18 hours of incubation. On day 3, the medium was changed to RPMI/B-27 without insulin and 5 M XAV939 (a Wnt/b-catenin inhibitor from Tocris Bioscience, Bristol, Bristol, UK). On day 5, the medium was changed to RPMI/B-27 without insulin. On day 7, the medium was changed to RPMI with 2% B-27 and insulin (Invitrogen, Carlsbad, CA, USA). On day 9, the medium was changed to RPMI 1640 medium without glucose and was supplemented with 2% B-27 with insulin. Then, using trypsin-ethylene diamine tetra-acetic acid (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), differentiated RUES2-cardiomyocytes (RUES2-CMs) were separated from the plate and re-plated on another plate.
RNA interference
The day before transfection, primary RUES2 were plated in six-well configurations. 50 nM of SOCS3, STAT3, and JAK siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were transfected into the cells in OPTI-MEM Medium (Invitrogen) for 48 hours using RNAiMAX (Invitrogen, Carlsbad, CA).
Animals
The Affiliated Cancer Hospital and Institute of Guangzhou Medical University laboratories were the source for the wild type C57BL/6 male mice (8 weeks old), which were handled in compliance with institutional and National Institutes of Health (NIH) standards. Before the trial, the mice spent a week getting used to their new habitat. The animals were kept in polypropylene cages with unlimited access to food and water. Under a 12 h light/12 h dark cycle, the room temperature was maintained between 18 and 22°C. To prevent any impacts of cohousing on the microbiota composition, the mice were housed separately in cages.
We first established a tumor-bearing mouse model. Mice were given 1 × 105 B16.F10 melanoma cells or PyMT breast cancer cells subcutaneously in the flask. Three treatment groups were assigned to B16-inoculated mice on 7 consecutive days and when the tumors grew to a size of around 100 mm3, it was determined that there was no statistically significant difference between the groups in tumor size (tumor size was calculated as follows: tumor volume (mm3) = long axis (mm) × short axis (mm) × short axis (mm) / 2.). Then the three groups received 1 ml intraperitoneal injections of phosphate-buffered saline (PBS) (sham group), 250 mg IgG (Bio X Cell, BE0090) (IgG group), or 250 mg BMS-1(PD-1/PD-L1 inhibitor 1) (Bio X Cell, BE0101) (BMS-1 group) every two days (10 mice per group). The mice were put to death 24 hours after the final BMS-1 injection, after which peripheral blood, tumors, and heart tissues were taken out and examined. An identical amount of phosphate buffer saline was intraperitoneally delivered into the mice in the control group (n = 10) (PBS group). The dosages of BMS-1 and IgG (10 mg/kg) utilized in this investigation were based on a report [25]. In the control group, wild-type C57BL/6 mice without B16.F10 melanoma cells received 1 ml intraperitoneal injections of PBS. Body weight and food intake were measured per animal.
Echocardiography
Mice were sedated with pentobarbital sodium (30 mg/kg) and put on a heating pad two weeks following the operation. Using Vevo770 (Visual Sonics Inc., Toronto, Canada) and a 716 probe, echocardiography was used to analyze the mice’s cardiac function in real time. For ventricular structure, transducers with a frequency of 17.5 MHz offered spatial resolutions. Ejection fraction (EF) and fractional shortening (FS) were deduced automatically by the High-Resolution Electrocardiograph system from the M-mode tracings, whereas left ventricular internal dimension in systole (LVIDs) and left ventricular internal dimension in diastole (LVIDd) were obtained from the M-mode tracings.
Pathological analysis
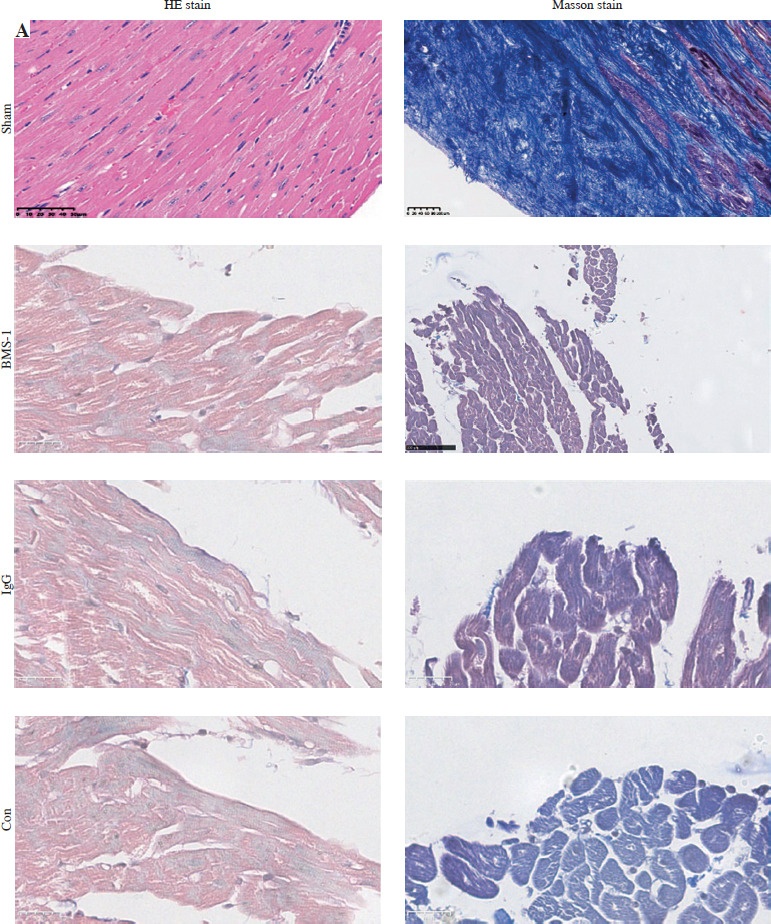
Mouse hearts were perfused at the time of scarification and fixed in 4% PFA for 24 h before the dehydration process. The dehydrated tissues were then paraffin-embedded and sliced into 5 µm sections using a microtome (Leica Rotary Microtome RM2235; Leica Biosystems, Buffalo Grove, IL). The sectioned tissues were incubated for 15 min at 65°C and then immersed in xylene for 5 min twice to remove paraffin. Samples were rehydrated by immersing in 100%, 95%, 80%, and 50% ethanol, each for 3 min. Samples were then stained three times with hematoxylin-eosin (H+E) and Masson’s trichrome for 5 min and eosin for 1 min for morphology observation. Slides from paraffin-embedded heart tissues were stained with Masson’s trichrome to detect fibrosis. Infarct size was evaluated as the average ratio of fibrosis area to the total ventricular area. Finally, we defined minimal inflammatory foci as inflammatory foci in the ventricle that constituted < 1% of the whole examined area to exclude cases with borderline/focal myocarditis resulting from local, systemic infection, or autoimmune mechanisms.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed as a measure of apoptosis using the DeadEnd fluorometric TUNEL system (Promega) according to the manufacturer’s protocol. For in vivo experiments, paraffin-embedded heart sections (5 µm) were fixed using 4% paraformaldehyde in phosphate-buffered saline for 1 h at room temperature, permeabilized with 0.5% Triton X-100 for 20 minutes, and then stained with TUNEL reagent containing fluorescein-12-dUTP to detect apoptotic cell nuclei and with 4′,6-diamidino-2-phenylindole (DAPI) to stain all cell nuclei. The sections were also co-stained with α-actinin (1 : 100; Cell Signaling Technology, #6487) and the associated Alexa Fluor 647-conjugated secondary antibody (1 : 200; Abcam, #ab150075) to confirm that apoptosis occurs in cardiomyocytes.
Real-time polymerase chain reaction (RT-PCR)
To isolate total RNA from ventricular tissue, Trizol reagent (Takara, Dalian, China) was used. By combining 50-100 mg of tissue with 1 ml of Trizol reagent and homogenizing, the tissue was instantly lysed using a homogenizer. The homogenized material was then mixed with 0.2 ml of chloroform, and it was let to sit for 20 minutes at room temperature. RNA was then precipitated by combining it with isopropyl alcohol. Using a microplate reader from Molecular Devices in Sunnyvale, California, the United States, total RNA yield was calculated at 260 nm. Then, using Oligo (dT) to separate the mRNA from the total RNA, it was reverse transcribed into first-strand complement DNA (cDNA), amplified, and then reconstituted using a Prime- Script 1st strand cDNA synthesis kit (Takara). 2 µl of cDNA, 12.5 µl of Takara’s SYBR Green 1 Master Mix, and 1 µl of each primer made up the reaction system. Using the iQ5 RT-PCR detection equipment, the PCR conditions were as follows: preincubation at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s and annealing/extension at 60°C for 30 s (Bio-Rad, Hercules, CA, USA).
Western blot
A rotor-stator homogenizer was used to homogenize the fresh ventricular tissue in ice-cold lysis buffer (Beyotime, Shanghai, China). After denaturation by boiling with loading buffer (Beyotime), SDS-PAGE gel was used to separate the proteins, which were then transferred to PVDF membrane. Samples were prepared under nonreducing, denaturing conditions and 6 µg of total protein was loaded into a gel (Bio-Rad, 4561084). Nonfat milk was used to block the membrane before primary antibodies for p-SOCS3 (PA5-86787), JAK (44-422G), p-STAT3 (12-9033-42), STAT3 (MA1-13042), VEGF (MA5-13182), and GAPDH (MA5-15738) were incubated at 4°C overnight, followed by washing. The membrane was incubated for an additional hour the next day using Cell Signaling’s HRP-conjugated secondary antibody. The bands detected by the FluorChem Image System were developed on the membrane using Immobilon solution (Millipore, Billerica, MA, USA).
Tissue preparation for flow cytometry
As previously mentioned, tumor tissues were converted to a single cell solution [26]. The following antibodies were used to immunostain the cells: CD45 Pacific Orange (MCD4530, Invitrogen), PD-L1 PE (12-5982-81, eBioscience), F4-80 APC (MCA497A647, Abd Serotec), CD86 PE (12-0862-83, eBioscience), Ly6C biotin (557359, BD Pharmingen) and Ly6G FITC (551460, BD Pharmingen). To ensure the specificity of immunostaining, cells were also stained with antibodies that were appropriately matched to their isotype.
Prior to staining in tumor tissue, macrophages were given a 4-hour treatment with the protein secretion inhibitor Brefeldin (BioLegend, 420601) to quantify intracellular cytokine production in the RUES2 cells. Macrophages were immunostained using the following antibodies: CD40 PE (eBioscience, 12-0409-42), CD86 FITC (BD Pharmingen, 560958), and CD14 APC (Bio-Rad, MCA1568A647). Additionally, matched isotype control antibodies were utilized to guarantee the immunostaining’s specificity.
Prior to immunostaining for IL12 PE (BD Pharmingen, 554479) and TNFa PE (eBioscience, MP6-XT22), the tumor sample was fixed with 4% PFA (Affymetrix, 19943) and permeabilized with 0.25 saponin in FACS buffer (1% BSA in PBS with 0.05% sodium azide) (eBioscience, 12-7321-81). mTOR (eBioscience, 2972S) and p-mTOR (eBioscience, 2971S) intracellular levels were stained using antibodies from Cell Signaling Technologies, and the primary antibody’s binding was detected using secondary donkey anti-rabbit IgG FITC (Jackson, 711-546-152).
Statistical analysis
The means and standard deviations (SD) of all values were reported. The statistical comparisons between groups were examined using one-way analysis of variance (ANOVA), such as in body weight and food intake. To assess the statistical significance of differences between two groups, a two-tailed Student’s t-test was utilized. A log-rank test was used to compare Kaplan-Meier survival curves. A difference was deemed statistically significant if the p value was less than 0.05.
Results
BMS-1 induces cardiotoxicity in mice with melanoma
We first analyzed the effects of BMS-1 intervention on body weight and intake in mice and found that BMS-1 significantly reduced body weight in mice 12 days after intervention relative to the control group. At the same time, there was no significant effect on the intake in mice (Fig. 1A, B).
Fig. 1
BMS-1 induces cardiotoxicity and impaired heart function in mice. A) Change of body weight. B) Food intake. C) Ratio of heart weight and body weight. D) Tumor weight. E) Representative figure of echocardiography. F) Fractional shortening indicators. G) Left ventricular internal dimension in systole. *p < 0.05, **p < 0.01. The sample size of each group is 10 H) Left ventricular internal dimension in diastole. I) Ejection fraction. J) BNP mRNA level. K) CK-MB. Values are presented as mean ± standard deviation. *p < 0.05, **p < 0.01. The sample size of each group is 10
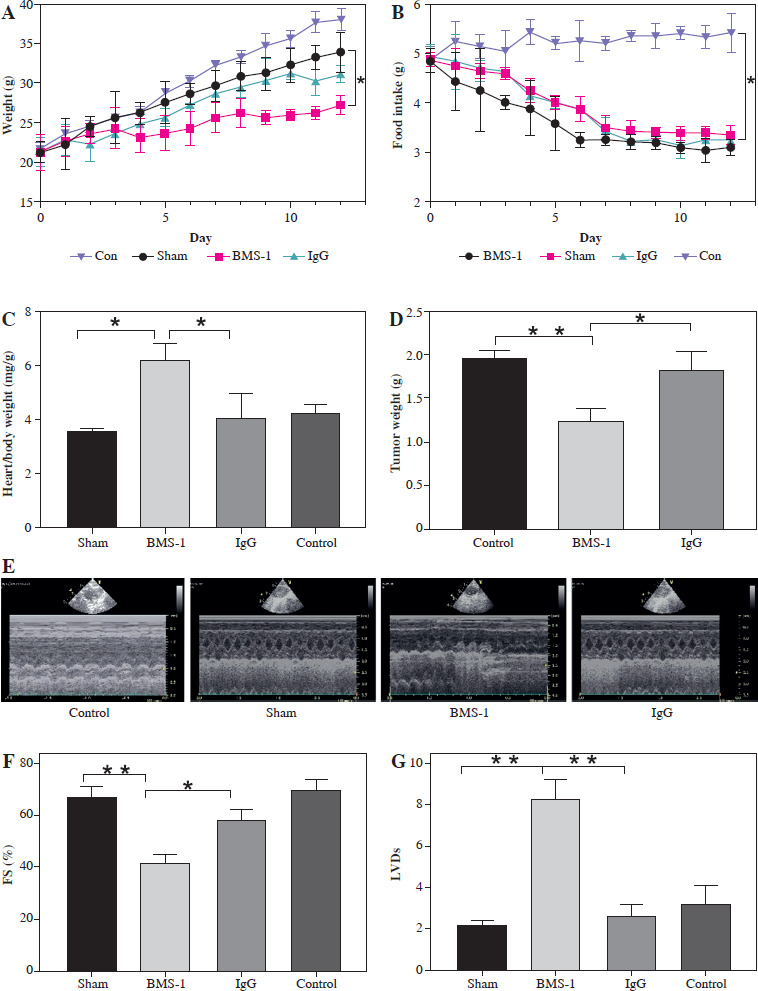
There was a significant increase in CK-MB, heart, and weight ratio after BMS-1 intervention relative to the control and IgG groups (Fig. 1).
To further clarify the changes in cardiac function, we used cardiac ultrasound to assess cardiac function in mice before execution and found that both fraction systolic index and ejection fraction were significantly lower in BMS-1 compared to the control and IgG groups, but left ventricular end-diastolic diameter and left ventricular end-systolic diameter were both elevated (Fig. 1F-I).
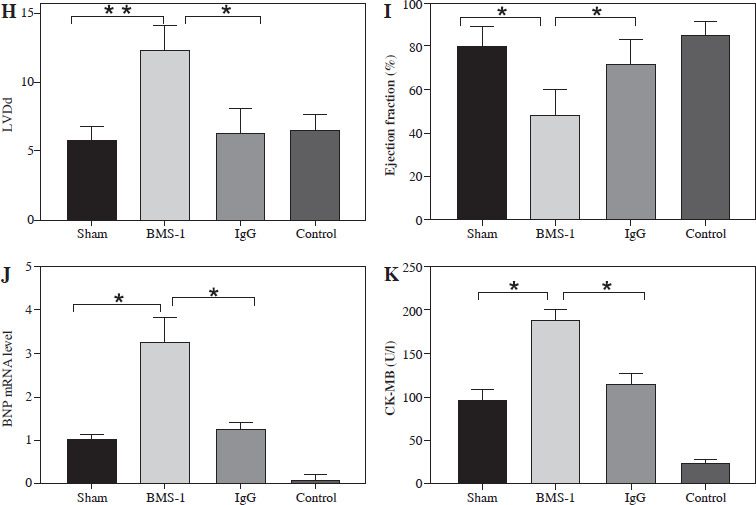
At the genetic level we analyzed the BNP mRNA levels in the myocardium and found that the BMS-1 group also showed significantly higher expression relative to the control and IgG groups. In addition, we stripped the tumor tissue intact and weighed the tumor tissue, and found that the tumor tissue weight of the mice showed a significant decrease after BMS-1 intervention. Finally, we used HE and Masson staining to analyze the damage of myocardial tissue by BMS-1, and the results showed a significant increase in the level of myocardial fibrosis in the BMS-1 group relative to the control and IgG groups. The above experimental results confirmed a significant improvement in tumor tissue weight in mice after BMS-1 intervention, as well as a significant effect of BMS-1 intervention on cardiac function in mice.
BMS-1 induces increased area of autophagy and regulates the SOC3/JAK/STAT3 signaling pathway
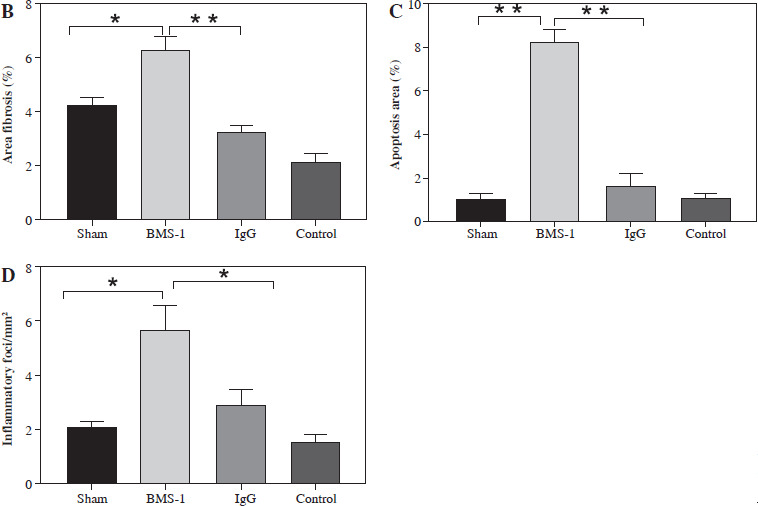
To investigate the possible mechanisms of PD-1 inhibitor-related myocardial toxic effects, we analyzed autophagy area and inflammatory status and found that BMS-1 was significantly elevated in the autophagic and inflammatory foci areas relative to the control and IgG groups. These results demonstrate that BMS-1-associated cardiotoxicity exhibits high levels of cellular autophagy and inflammation (Fig. 2).
Fig. 2
BMS-1 induces myocardial fibrosis and autophagy in mice. A) Representative images of hematoxylin and eosin (HE) staining and Masson staining of cardiac tissues and corresponding quantification analysis B) Area of fibrosis. C) Apoptosis area. D) Inflammatory foci. Values are presented as mean ± standard deviation. *p < 0.05, **p < 0.01. The sample size of each group is 10
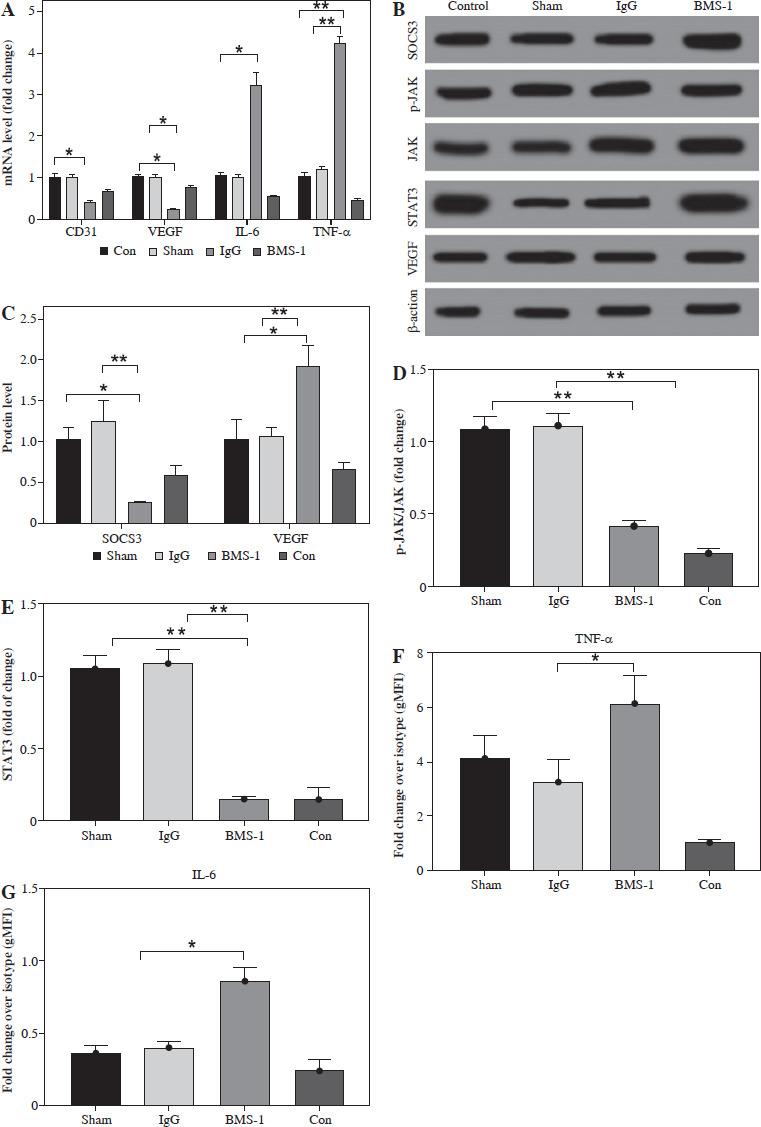
We also analyzed angiogenesis-related genes and inflammation genes. The results indicated that BMS-1 showed significant downregulation of CD31 and VEGF but significant upregulation of IL-6 and TNF-α levels relative to the control group (Fig. 3A). At the protein level, we further analyzed the effect of the SOCS3 signaling pathway in BMS-1 intervention in cardiomyocytes. The results showed a significant decrease in BMS-1 relative to SOCS3, p-JAK/JAK and STAT3 (Fig. 3B-E). However, VEGF levels appeared to be significantly upregulated. This also corresponds to the gene level. In addition, PD-L1 antibody-treated macrophages also increased production of TNF-α and IL-12, consistent with PD-L1 antibody treatment inducing production of inflammatory macrophages (Fig. 3F, G).
Fig. 3
BMS-1 induced SOCS3/JAL/STAT3 signaling pathway and VEGF expression in RUES2 cells. A) mRNA level in heart tissue. B) Representative images of WB. C) SOCS3 and VEGF protein level. D) p-JAK/JAK protein level. E) STAT3 protein level. F) TNF-α level. G) IL-6 level. Values are presented as mean ± standard deviation. *p < 0.05, **p < 0.01. Each group of experiments was repeated three times
Effect of silencing SOCS3, JAK and STAT3 on SOC3/JAK/STAT3 signaling pathways in BMS-1 induces myocardial injury
To further analyze the effect of PD-1 inhibitors on the myocardium, we silenced SOCS3, JAK and STAT3 in the signaling pathway, and the results showed that SOCS protein levels and VEGF levels were significantly downregulated after SOCS3 silencing (Fig. 4A-C).
Fig. 4
Effect of silencing SOCS3, JAK and STAT3 on SOC3/JAK/STAT3 signaling pathways in BMS-1 induces myocardial injury. A) Representative images in silencing SOCS3, JAK and STAT3 experiment. B) SOCS3 and VEGF protein level. C) p-JAK/JAK protein level. D) p-STAT3/STAT3 protein level. Values are presented as mean ± standard deviation. *p < 0.05, **p < 0.01. Each group of experiments was repeated three times
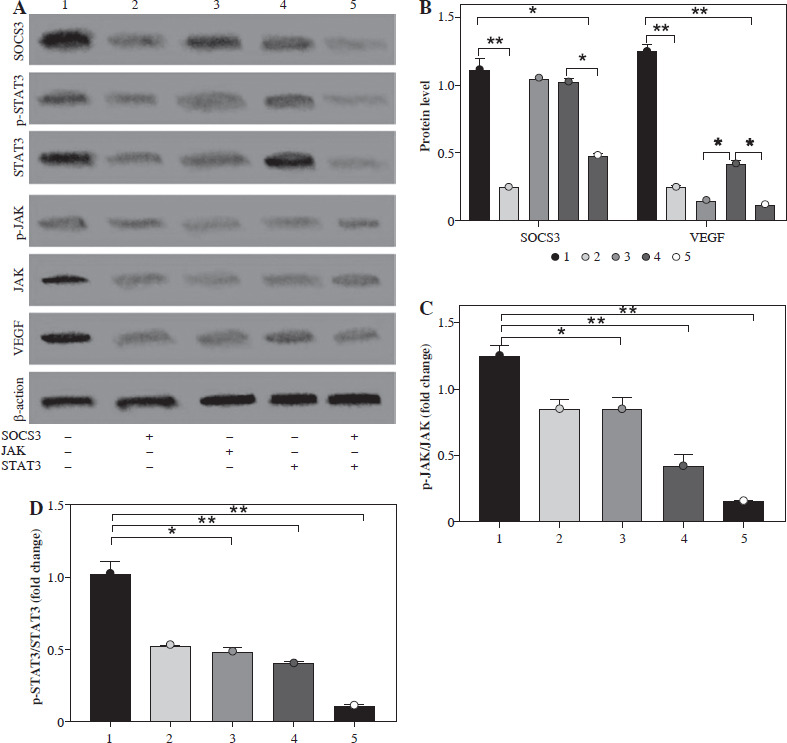
Also, significant downregulation of p-JAK/JAK and p-STAT3/STAT3 levels was found after silencing of JAK and STAT3. However, the effect of silencing STAT3 on JAK levels was not significant. These results suggest that SOC3/JAK/STAT3 signaling pathways may play an important role in BMS-1 intervention in myocardial injury (Fig. 4).
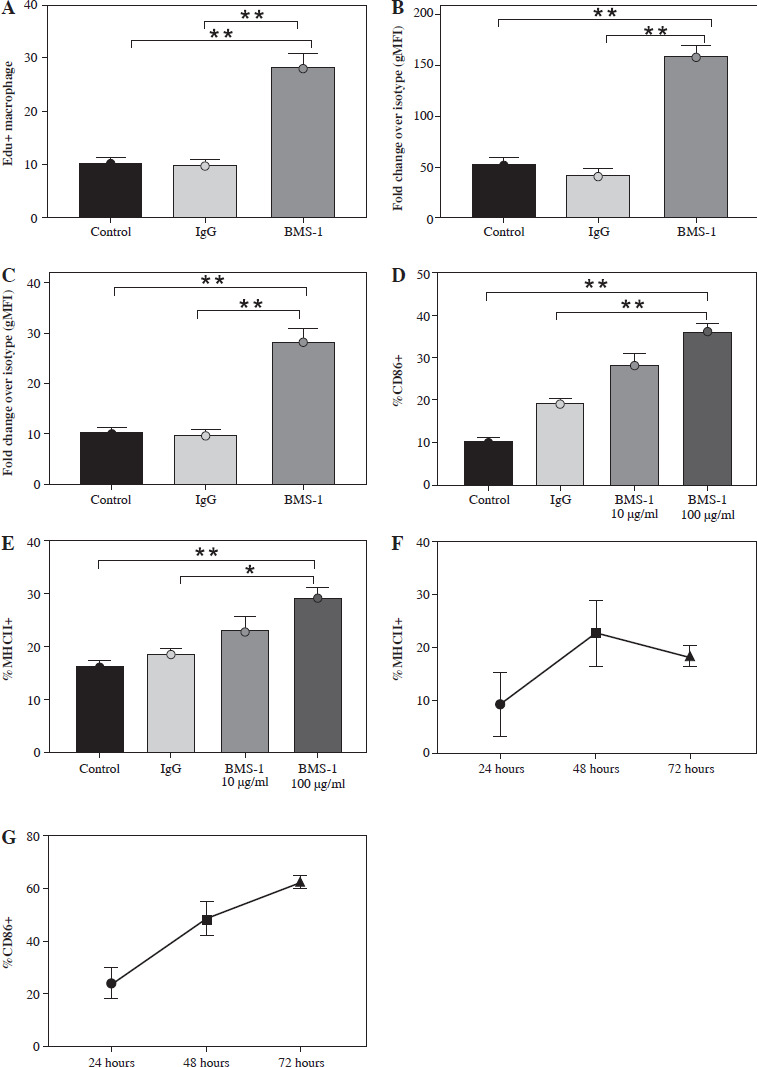
BMS-1 treatment induces macrophage activation
We then analyzed whether the PD-1 inhibitor BMS-1 activates macrophages after induction. BMS-1 treatment significantly upregulated the costimulatory molecules CD86 and MHC II relative to the control and IgG groups, which was also consistent with the macrophage activation state. This effect was titratable with increasing concentrations of PD-L1 antibody. Macrophage changes were first apparent after 48 hours of treatment (Fig. 5). These results suggest that PD-1 antibodies may induce macrophage polarization.
Fig. 5
BMS-1 treatment induced macrophage activation in tumor tissue. Macrophages were treated with medium only, IgG, or BMS-1 for 72 hours with different concentrations (BMS-1 10, 100 μg/ml). A) Proliferation was measured using EdU incorporation. B, C) They were stained to evaluate costimulatory molecule expression (CD86 and MHCII) by flow cytometry, and the geometric mean fluorescence intensity (gMFI) is shown as fold increase over an irrelevant isotype stain. D-G) BMS-1 was titrated (E) and macrophages were harvested at different time points following treatment with 100 mg/ml BMS-1. Values are presented as mean ± standard deviation. *p < 0.05, **p < 0.01. Each group of experiments was repeated three times
Discussion
PD-1/PD-L1 acts as a co-inhibitory checkpoint molecule to suppress T cell responses as the primary mechanism. Previous studies have found that PD-1 antibody treatment significantly inhibits tumor regression, which also indicates the anti-tumor effect of PD-1 antibodies. However, the adverse cardiac side effects associated with PD-1 antibodies are also attracting increasing attention [27-29]. Key findings from our study found that PD-1 anti- bodies can reduce tumor volume but inflict significant damage to cardiomyocytes and heart function. The possible mechanism is related to macrophage polarization and the SOC3/JAK/STAT3 signaling pathway.
The immune system can be modulated to prevent auto- immune reactions brought on by excessive activation of immune cells by immune checkpoint molecules that serve as protective factors for the human immune system, such as cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and PD-1/PD-L1 [30, 31]. Immunosuppression increased the risk of myocarditis 11-fold compared to controls [32]. The greatest known risk factor for ICI-associated myocarditis is receiving combination ICI therapy, such as CTLA-4 inhibitor in conjunction with PD-1 inhibitor [33, 34]. Myocarditis following ICI therapy may be more frequent than is thought, manifests quickly after commencing treatment, has a malignant course, and responds to increased steroid dosages, according to research by Mahmood et al. [35]. In the aforementioned retrospective pharmacovigilance investigation, 50% of ICI-associated myocarditis cases resulted in death [32]. When treating myocarditis cases, ICI combination therapy was more frequently deadly (65.6%) than ICI monotherapy (44.4%). Atrial and ventricular arrhythmias, as well as heart block, are at an increased risk, according to preliminary findings. Additionally, only about 50% of the patients show signs of a decreased ejection fraction [7, 35]. Serum cardiac biomarkers including cardiac troponin and creatine kinase-muscle/brain (CK-MB) are virtually invariably high in ICI-associated myocarditis, although their individual positive predictive values vary. Cardiovascular magnetic resonance imaging and most notably endomyocardial biopsy are frequently used to detect myocardial inflammation. Early histological analysis of the heart, the cardiac conduction system, and skeletal muscle indicated infiltration of numerous CD4+ and CD8+ T cells and CD68+ macrophages [36, 37]. In our study, significant cardiac impairment was also discovered when immunosuppressive medications such as BMS-1 were administered to melanoma mice.
The JAK-STAT signaling pathway’s crucial attenuator, suppressor of cytokine signaling 3 (SOCS3), is involved in regulating the strength and duration of STAT3 activation during the process of myocardial protection [38]. Cytokines that activate JAK-STAT3 have been shown to protect the heart. The cardiac-specific deletion of SOCS3 reduces left ventricular remodeling following myocardial infarction (MI), decreases myocardial apoptosis and fibrosis, and increases the activation of cardioprotective signaling pathways [39, 40]. According to recent research, SOCS proteins modify the activity of activated STATs, which changes the activation of downstream genes [41]. This adds diversity to the JAK-STAT pathway. STAT3 is crucial for the transcription of PD-L1 when it is activated by different pathways [42]. According to Wu et al., melanoma with high PD-L1 expression is resistant to anti-PD-1 therapy when SOX2-JAK-STAT signaling is targeted [43]. In contrast, STAT3 silencing and inhibition lowered PD-L1 expression in STAT3 mutant NKTL cell lines. [44]. Zhang et al. discovered that STAT3 activation confers high PD-L1 expression, which may facilitate tumor immune evasion. Granulocyte colony stimulating factor has the ability to bind to the granulocyte colony stimulating factor receptor and control the SOCS3-JAK-STAT3 pathway, which in turn regulates the infiltration of neutrophils and macrophages into the liver [15]. Ge et al. discovered that SOCS3/JAK/STAT3 regulation may have a potentially harmful impact in the progression of B[a]P-induced hepatocellular carcinoma [14]. Immunosuppression significantly affected the SOCS3/JAK/STAT3 signaling pathway, which is consistent with our findings.
Resetting tumor-associated macrophages (TAMs) is a promising method to improve tumor immunosuppression, boost T-cell antitumor immunity, and improve the effectiveness of anti-PD-1 antibody therapy and long-term memory immunity. In mouse cancer models throughout time and with increasing disease stage in primary human malignancies, TAM PD-1 expression rises [45]. In mouse cancer models, inhibition of PD-1-PD-L1 in vivo improves macrophage phagocytosis, slows tumor growth, and lengthens mouse survival in a macrophage-dependent manner [46]. Tumor-associated macrophage PD-1 expression correlates adversely with phagocytic potency against tumor cells according to Liu et al. [47]. Anti-PD-1/PD-L1 therapeutic resistance was eventually brought on by exosomal PD-L1-induced macrophage M2 polarization. Major histocompatibility complex (MHC) class II positivity on tumor cells is related to treatment response, progression-free and overall survival, as well as CD4(+) and CD8(+) tumor infiltration in melanoma patients receiving anti-PD-1 therapy. To enhance anti-PD-1 patient selection, MHC-II(+) tumors can be recognized by melanoma-specific immunohistochemistry utilizing commercially available HLA-DR antibodies [48]. MHC class I molecule downregulation is one of the most common ways for tumors to evade the immune system of the host; hence it should be taken into account when PD-1/PD-L1 checkpoint inhibition is being used [49]. In addition, the use of MHC class I as a predictive biomarker should be taken into account because its loss may pose a challenge to successfully enhancing the antitumor adaptive immune response through PD-1/PD-L1 inhibition [50]. Additionally, these findings imply that, in addition to the immune response-based mechanism, the anticancer impact of BMS may also be partially mediated by a direct off-target cytotoxic effect. A valuable approach for the screening of small molecule inhibitors of PD-1/PD-L1 binding that can prevent tumor growth via an immune response-mediated mechanism has also been demonstrated for the humanized dKO NOG mouse model used in this study [51]. The polarization of immune cells was found to be positively affected by immunosuppression in our study, which may also be a mechanism for the effect of enhanced cardiac function.
Limitations
This study made some significant discoveries, yet there are still certain limitations. This study primarily examined PD-1/PD-L1 inhibitor-associated myocarditis; more research into other types of ICI-related cardiotoxicity is required. In addition, only mice treated with BMS-1 were used to study the mechanism of PD-1/PD-L1 inhibitor-related cardiotoxicity. To confirm these findings, additional studies using animals and people treated with anti-PD-1/PD-L1 antibodies and larger sample sizes will be carried out.
Conclusions
Our findings firstly demonstrate that there are two possible mechanisms by which PD-1/PD-L1 inhibitors cause cardiotoxicity and aberrant cardiac function. First, PD-1/PD-L1 inhibitors initiated a SOCS3/JAK/STAT3 negative feedback loop, which may have contributed to the development of cardiotoxicity. Second, the synthesis and mobilization of macrophages are controlled by PD-1/PD-L1 inhibitors, which indirectly affect the development of cardiotoxicity. This research suggests a potential method by which PD-1/PD-L1 inhibitors may result in cardiotoxic effects.