
Introduction
Lung adenocarcinoma (LUAD), the most common subtype of lung cancer, mainly arises from the epithelium of the bronchial mucosa, with some cases originating from the mucous glands of the larger bronchi. It is distinguished by its highly infiltrative and destructive growth pattern [1]. Surgical intervention is the preferred choice for early-stage LUAD patients, while patients in the advanced stages also require supplementary treatments including radiotherapy, chemotherapy, and targeted medication [2]. Patients with LUAD exhibit varied and subtle symptoms, often diagnosed late, resulting in a poor prognosis. How to improve the current survival outcomes for LUAD patients remains a daunting challenge [3]. Anti-angiogenic targeted therapy has been widely adopted in clinical settings recently, proving effective for treating advanced or metastatic LUAD [4]. It is reported that angiogenesis within the tumor microenvironment (TME) is a necessary step for tumor invasion and metastasis and feeds into the progression of malignancy [5]. Thus, a thorough understanding of the molecular mechanisms of angiogenesis in LUAD and the discovery of key driver genes could lead to new therapeutic targets.
The genesis of tumors is contingent upon the intrinsic qualities of the cancer cells as well as their interactions with the components of the TME [6]. Tumor-associated macrophages (TAMs), which are among the most populous immune cells within the TME, are highly malleable and can polarize into either the M1 or M2 phenotype in reaction to different stimuli, showing a range of functions. M1-type macrophages are known to release pro-inflammatory cytokines that provoke a sustained inflammatory response and are instrumental in activating the immune system by modulating the activity of T lymphocytes (T cells) or natural killer (NK) cells [7]. M2 macrophages induced by (T helper 2) Th2 cell cytokines such as interleukin (IL)-4 and IL-13 can secrete anti-inflammatory factors such as IL-10 and tumor necrosis factor β (TNF-β), recruit Th2, regulatory T cells, etc., and initiate functional anti-inflammatory regulatory mechanisms [8]. Haydar et al. [9] reported that azithromycin can effectively manage hyperinflammation in patients with cystic fibrosis by suppressing the STAT1 and NF-κB signaling pathways, leading to the polarization of macrophages into the M2 type. Beyond their involvement in inflammatory conditions, the role of macrophages within the TME in cancer treatment has been increasingly scrutinized. Many studies have highlighted that M2-polarized macrophages contribute to the progression of various malignancies by generating a substantial amount of growth factors, molecules involved in extracellular matrix remodeling, and cytokines, which promote tumorigenesis, stimulate angiogenesis, and inhibit anti-tumor immune responses, as seen in hepatic, colorectal, and pancreatic cancers [10]. Angiogenesis is essential for tumor growth and metastasis, with M2-polarized TAMs being capable of expressing multiple angiogenic factors, including vascular endothelial growth factor and matrix metalloproteinase 9, which facilitate the formation of new blood vessels [11]. Research into the interaction between LUAD cells and TAMs has identified high levels of SPP1 expression in both tumor tissues and macrophages. Elevated SPP1 expression can increase the expression of PD-L1, thus promoting the M2 polarization of macrophages, indicating that SPP1 might be a potential therapeutic target for M2 macrophage polarization and LUAD treatment [12]. An in-depth exploration of the mechanisms behind macrophage polarization within the TME and the identification of additional molecular targets that control M2 polarization will pave the way for greater improvement in LUAD treatment.
The ankyrin repeat domain (ANKRD) constitutes a protein motif that is prevalent in the natural world, existing in both eukaryotic and prokaryotic organisms as well as viruses. The ANKRD family is large and is involved in a wide array of physiological processes within the body, including control of the cell cycle, transcription, cell signaling, cell apoptosis, and inflammatory reactions [13]. Ankyrin repeat domain-containing protein 22 (ANKRD22), a member of the ANKRD family, has four ankyrin repeat units and contains 191 amino acids, intimately connected with the genesis and progression of various tumors [14]. ANKRD22 expression is mainly detected in normal gastrointestinal epithelial cells, macrophages, and tumor cells [15]. Silencing ANKRD22 in breast cancer (BC) has been shown to suppress the proliferation, invasion, and epithelial-mesenchymal transition (EMT) of malignant cells. Mechanistically, the knockdown of ANKRD22 interrupts the Wnt/β-catenin signaling cascade by reducing the expression of NuSAP1, which in turn inhibits the malignant behavior of BC cells. Thus, ANKRD22 could be a valuable diagnostic biomarker for BC [16]. Yin et al. [17] discovered through an analysis of clinical relevance that ANKRD22 is overexpressed in primary non-small cell lung cancer (NSCLC) tissues compared to the adjacent tissues. The elevated expression of ANKRD22 correlates markedly with shorter relapse and overall survival periods for NSCLC patients. The investigation into the molecular mechanisms by which ANKRD22 might regulate the progression of tumors, particularly in LUAD, is not yet comprehensive. Therefore, it is imperative to further examine the regulatory mechanisms of ANKRD22 in relation to the malignant behavior of LUAD.
In our study, we observed that ANKRD22 was overexpressed in LUAD tissues and cell lines, and this overexpression could exacerbate the malignancy of LUAD cells by promoting the M2 polarization of macrophages (derived from THP-1 cells).
Material and methods
Bioinformatics analysis
Utilizing the edgeR R package, differential expression analysis (|logFC| > 1.0, FDR < 0.05) was performed on the LUAD mRNA expression data (normal: 59 samples, tumor: 539 samples) obtained from the TCGA database. This analysis yielded a set of differentially expressed mRNAs. After consulting the literature, the target gene for this study was established and its expression levels in both normal and tumor samples were assessed.
Cell culture
The human lung epithelial cell line BEAS-2B (BNCC359274), LUAD cell lines A549 (BNCC337696), H1975 (BNCC340345), Calu-3 (BNCC359757), human acute monocytic leukemia cell line THP-1 (BNCC358410), and human umbilical vein endothelial cells (HUVEC) (BNCC342438) were all procured from BNCC (China). All these cell lines are verified with an STR profiling report and are routinely screened to ensure there is no mycoplasma contamination. BEAS-2B cells were cultured in DMEM-H complete growth medium (BNCC338068), A549 cells in F-12K complete growth medium (BNCC338550), H1975 in RPMI-1640 complete growth medium (BNCC338360), Calu-3 in MEM complete growth medium (BNCC338137), THP-1 cells in THP-1 specific culture medium (BNCC354257), and HUVEC cells in HUVEC specific culture medium (BNCC360874). These media were all supplemented with 10% FBS, also obtained from BNCC (China). 1% penicillin-streptomycin (Sigma, USA) was added to the growth medium, and the cells were cultivated in a 37°C incubator with 5% CO2. The cell populations used in all experiments were between the 5th and 10th passages, and the cells were in a healthy state.
The induction method for M0 macrophages was referenced from a previous study [18]. THP-1 cells in good growth condition were selected and treated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma, USA) for 24 h. Subsequently, the cell status was observed every 6 h. When all cells transitioned from suspension growth to adherent growth, changed from round to irregular morphology, and increased in cell volume, it was determined that THP-1 cells had been successfully induced into macrophages.
Cell transfection
The sequences for si-NC and si-ANKRD22 were designed and created by RiboBio (China). For the overexpression of ANKRD22, the pcDNA3.1 plasmid was used to construct the expression vector. The ANKRD22 sequence, synthesized by GenePharma (China), was cloned into the pcDNA3.1 to form the oe-ANKRD22 plasmid, with the empty pcDNA3.1 plasmid serving as a control (oe-NC). Lipofectamine 2000 (Invitrogen, USA) was used as per the manufacturer’s guidelines to transfect the A549 cells with both the oe-NC and oe-ANKRD22 plasmids along with siRNAs. The cells were then collected for subsequent analysis 48 h following transfection.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Cells were washed twice with pre-cooled phosphate-buffered saline (PBS) and then collected. RNA was extracted using Trizol reagent (Thermo Fisher, USA), and the RNA concentration and quality were measured using a nucleic acid and protein quantification instrument. The RNA was reverse-transcribed into cDNA with the aid of the PrimeScript RT Reagent Kit (Takara, Japan), paving the way for subsequent qPCR utilizing the SYBR Green Realtime PCR Master Mix (Toyobo, Japan). The PCR conditions were as follows: 95°C for 60 seconds, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. To ascertain the relative expression levels of the target genes, a comparison was made against the GAPDH reference gene, employing the widely accepted 2-ΔΔCT method for quantification. The qRT-PCR primers were all procured from You Kang Biological (China), with the respective sequences for each primer detailed in Table 1.
Table 1
Primer sequences
Apoptosis evaluation
The Cell Counting Kit-8 (CCK-8) kit (MCE, USA) was utilized to test A549 cell viability. Transfected cells were plated at a density of 5 × 103 cells per well in a 96-well plate, with triplicate wells for each experimental condition. The plate was incubated for 48 h in a 37°C incubator with 5% CO2. Finally, 10 µl of CCK-8 solution was added to each well for a 2-h incubation. The optical density at 450 nm was then read using a microplate spectrophotometer.
Western blot
Total cell proteins were extracted with RIPA lysis buffer (Beyotime, China), and protein concentrations were assessed using a BCA protein assay kit (Beyotime, China). With the loading buffer (Solarbio, China) mixed into the samples, a denaturation process at 100°C for 10 min was initiated in a metal bath. Following this, the proteins were resolved by SDS-PAGE and then electrophoretically transferred to a 0.45 µm polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The membrane underwent a blocking step with 5% skim milk powder for 1 h to prevent non-specific antibody binding. Primary antibodies specific for CD80, CD163, and GAPDH (1 : 1000 for CD80 and CD163, 1 : 10000 for GAPDH, all from Abcam, UK) were applied and incubated overnight at 4°C. The membrane was subsequently washed three times with TBST for 5 min each to remove excess primary antibodies. Finally, a secondary goat anti-rabbit IgG H&L (HRP) antibody (1 : 5000, Abcam, UK) was applied for 1 h at room temperature to facilitate detection. After another three washes for 5 min each in TBST, the proteins were visualized using an ECL detection kit (ABclonal, China) and a chemiluminescence imaging system.
Flow cytometry
Following transfection, A549 cells were cultured for 24 h, after which the supernatant was centrifuged at 3000 rpm for 10 min to pellet any cells. The supernatant after centrifugation was utilized to treat M0 macrophages for 48 h. The macrophages were then dissociated into a single-cell suspension using trypsin. The cells were incubated with PerCP Anti-Human CD68 Antibody (1 : 100; 333813, Bio- Legend, USA), PE Anti-Human CD86 Antibody (1 : 100; E-AB-F1012D, Elabscience, China), and/or FITC Anti- Human CD206/MMR Antibody (1 : 100; E-AB-F1161C, Elabscience, China) for 15 min. After washing with PBS, the cells were resuspended, and the flow cytometry data were collected and analyzed using Agilent’s flow analysis software. The selection of M1/M2 macrophage markers was referenced from a previous study [19]. Among them, CD68 is a general marker for macrophages, while CD86 corresponds to M1 macrophages, and CD206 corresponds to M2 macrophages.
Colony formation assay
Upon the completion of transfection, A549 cells were evenly distributed at a rate of 200 per well across a 12-well plate and allowed to grow in a 37°C, 5% CO2 incubator until colonies became visible. After a wash with PBS, the cells were fixed with 75% ethanol and stained with 1% crystal violet (Sigma, USA) to observe colony formation. Colony counting was conducted using Image-Pro Plus software (USA).
Transwell assay
Transwell experiments were performed in 24-well plates (Corning, USA). A549/HUVEC cells were resuspended in medium without serum and placed into the upper chamber at a rate of 1 × 104 cells per well, while the lower chamber contained medium with 10% FBS. After 48 or 72 h of culture at 37°C, the Transwell inserts were extracted, and the cells remaining in the upper chamber were carefully scraped off with a cotton-tipped swab. The cells were fixed with 75% ethanol for 30 min and stained with 0.1% crystal violet for 20 min, then washed twice with PBS. Once dried, the cells were imaged under a microscope and counted using Image-Pro Plus software (USA).
Angiogenesis experiment
Matrigel (BD Biosciences, USA) was thawed at 4°C, mixed evenly with a pre-cooled pipette tip, and then transferred to a 96-well plate. The plate was incubated at 37°C for 1 h for the Matrigel to gel. Thereafter, treated HUVEC cells were digested, centrifuged, counted, and resuspended in a culture medium before being plated at a density of 1 × 104 cells per well on the Matrigel. The plate was then incubated for 6 h in a 37°C, 5% CO2 incubator. Angiogenesis was observed and photographed under a microscope, and data analysis was facilitated by ImageJ software.
Statistical analysis
The experimental data were reported as the mean ± SD, with triplicate execution for each to ensure accuracy. GraphPad Prism 8.0 (USA) was used for statistical analysis, with t-tests to compare two groups and one-way ANOVA for comparisons across multiple groups. Statistical significance was defined by p < 0.05.
Results
ANKRD22 is highly expressed in LUAD
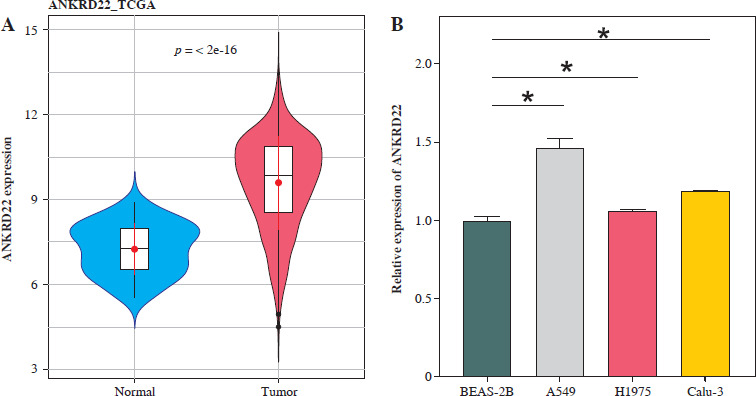
To determine the expression levels of the ANKRD22 gene in LUAD, a primary assessment was executed using the TCGA-LUAD database, focusing on its expression within LUAD tissues. The findings revealed that the expression levels of ANKRD22 were markedly elevated in LUAD tissues compared to normal tissues (Fig. 1A). Subsequently, qRT-PCR was employed to assess the mRNA expression levels of ANKRD22 in human lung epithelial cells (BEAS-2B) and LUAD cell lines (A549, H1975, Calu-3). The data showed that the relative mRNA expression levels of ANKRD22 were notably higher in A549, H1975, and Calu-3 cells than in BEAS-2B cells (Fig. 1B). Collectively, ANKRD22 expression was upregulated in both LUAD tissues and cell lines.
Fig. 1
Elevated expression of ANKRD22 in lung adenocarcinoma (LUAD). A) Depiction of expression levels of ANKRD22 within LUAD tissues, as analyzed via the TCGA-LUAD database, with the blue color symbolizing normal tissue samples (n = 59) and red symbolizing tumor tissue samples (n = 539); B) qRT-PCR was used to measured the expression levels of ANKRD22 in human lung epithelial cells (BEAS-2B) and LUAD cell lines (A549, H1975, Calu-3). All experiments were conducted with 3 independent replicates. The data are presented as mean ± standard deviation. *p < 0.05
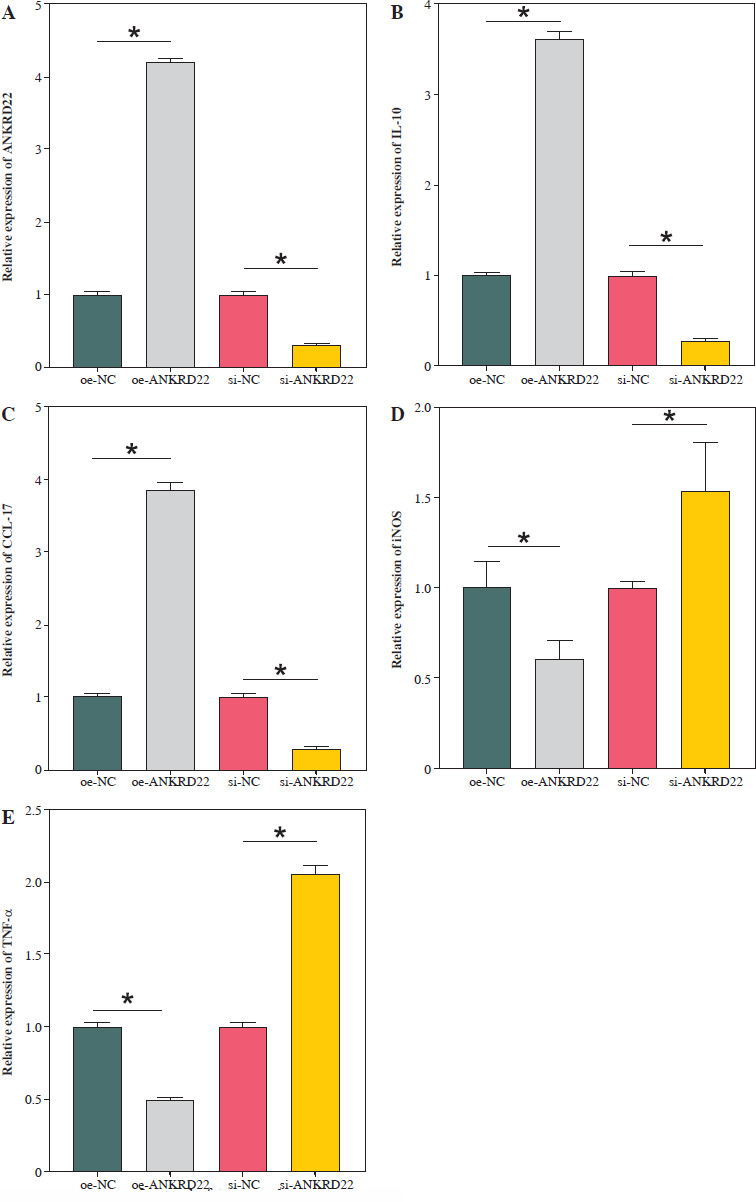
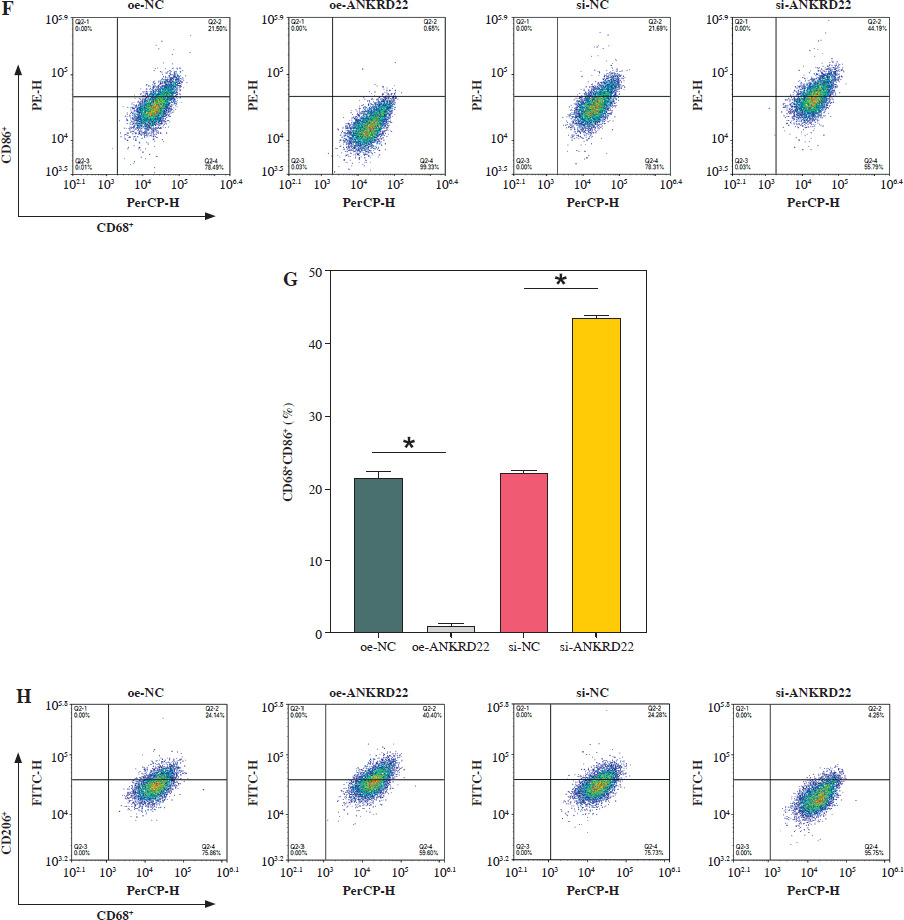
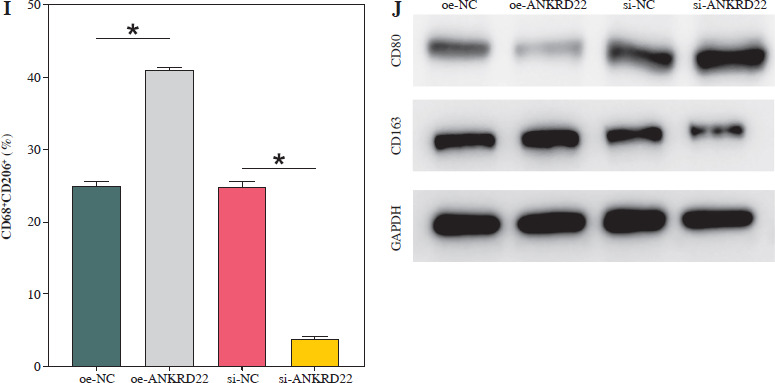
ANKRD22 promotes macrophage M2 polarization
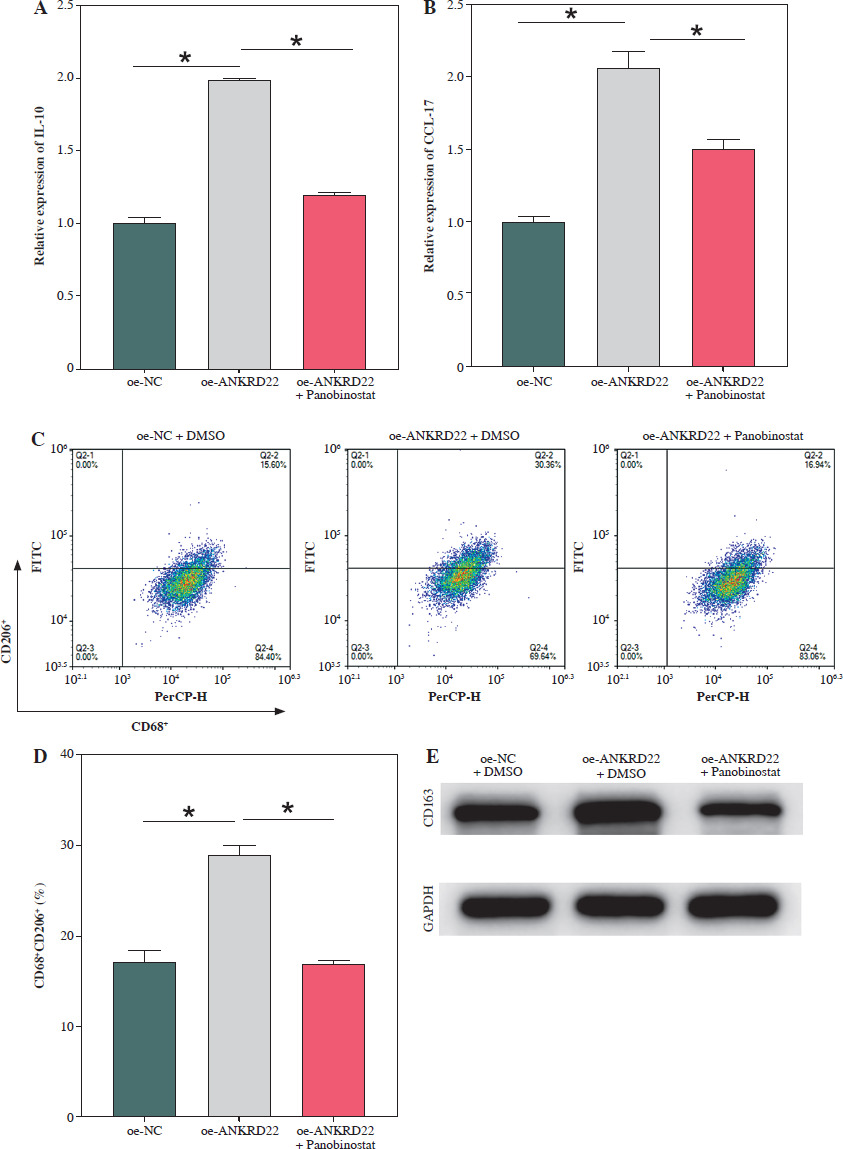
It has been documented that ANKRD22 participates in the regulation of TME in a multitude of cancers, potentially being associated with the polarization of TAMs. To ascertain the influence of ANKRD22 expression levels on macrophages within LUAD, we performed overexpression or knockdown of ANKRD22 in A549 cells, categorizing them into the following groups: oe-NC, oe-ANKRD22; si-NC, si-ANKRD22. The efficiency of transfection was evaluated by qRT-PCR (Fig. 2A). The supernatants from the culture medium of these categorized cells were harvested to cultivate THP-1 cells which had been stimulated with PMA. The expression levels of M1 and M2 macrophage markers were subsequently detected to delineate the effect of ANKRD22 on macrophage polarization. qRT-PCR results showed that overexpression of ANKRD22 significantly upregulated the mRNA relative expression levels of M2 macrophage markers IL-10 and CCL-17, while the opposite was observed upon ANKRD22 knockdown (Fig. 2B, C). When the oe-ANKRD22 group was compared to the oe-NC group, markedly lower mRNA expression levels of the M1 macrophage markers iNOS and tumor necrosis factor α (TNF-α) were observed. In contrast, the knockdown of ANKRD22 (si-ANKRD22 group) led to significantly higher levels of these markers compared to the si-NC group (Fig. 2D, E). These collective results suggested that the elevated expression of ANKRD22 could suppress the polarization of M1 macrophages and enhance the polarization of M2 macrophages. Subsequent flow cytometry to determine the proportions of M1 (CD68+CD86+) and M2 (CD68+CD206+) macrophages confirmed that the overexpression of ANKRD22 led to a pronounced reduction in the proportion of CD68+CD86+ macrophages (Fig. 2F, G) and a corresponding increase in the proportion of CD68+CD206+ macrophages (Fig. 2H, I), with inverse results observed upon the knockdown of ANKRD22. Furthermore, the expression of CD80, a protein marker for M1 macrophages, and CD163, a protein marker for M2 macrophages, was assessed via Western blot (WB). As depicted in Figure 2J, relative to the oe-NC group, the oe-ANKRD22 group displayed markedly lower in CD80 expression and markedly higher CD163 expression. The si-ANKRD22 group exhibited much higher CD80 and lower CD163 expression levels when compared to the si-NC group. In conclusion, the expression levels of ANKRD22 were capable of affecting macrophage polarization in LUAD, with increased expression favoring M2 polarization and decreased expression inhibiting it.
Fig. 2
Elevated expression of ANKRD22 promotes M2 macrophage polarization. A) qRT-PCR was performed to detect transfection efficiency in oe-NC/oe-ANKRD22 and si-NC/si-ANKRD22. B-E) qRT-PCR was used to measure mRNA expression levels of M2 macrophage markers (IL-10, CCL-17) and M1 macrophage markers (iNOS, TNF-α) in A549 cells. The data are presented as mean ± standard deviation. *p < 0.05 F-H) Flow cytometry was used to detect the effects of ANKRD22 overexpression or knockdown on the proportions of CD68+CD86+ and CD68+CD206+ macrophages. All experiments were conducted with 3 independent replicates. The data are presented as mean ± standard deviation. *p < 0.05 I) Flow cytometry was used to detect the effects of ANKRD22 overexpression or knockdown on the proportions of CD68+CD86+ and CD68+CD206+ macrophages. J) WB was used to detect the effects of ANKRD22 overexpression or knockdown on the protein expression levels of CD80 and CD163. All experiments were conducted with 3 independent replicates. The data are presented as mean ± standard deviation. *p < 0.05
ANKRD22 encourages LUAD malignant behaviors
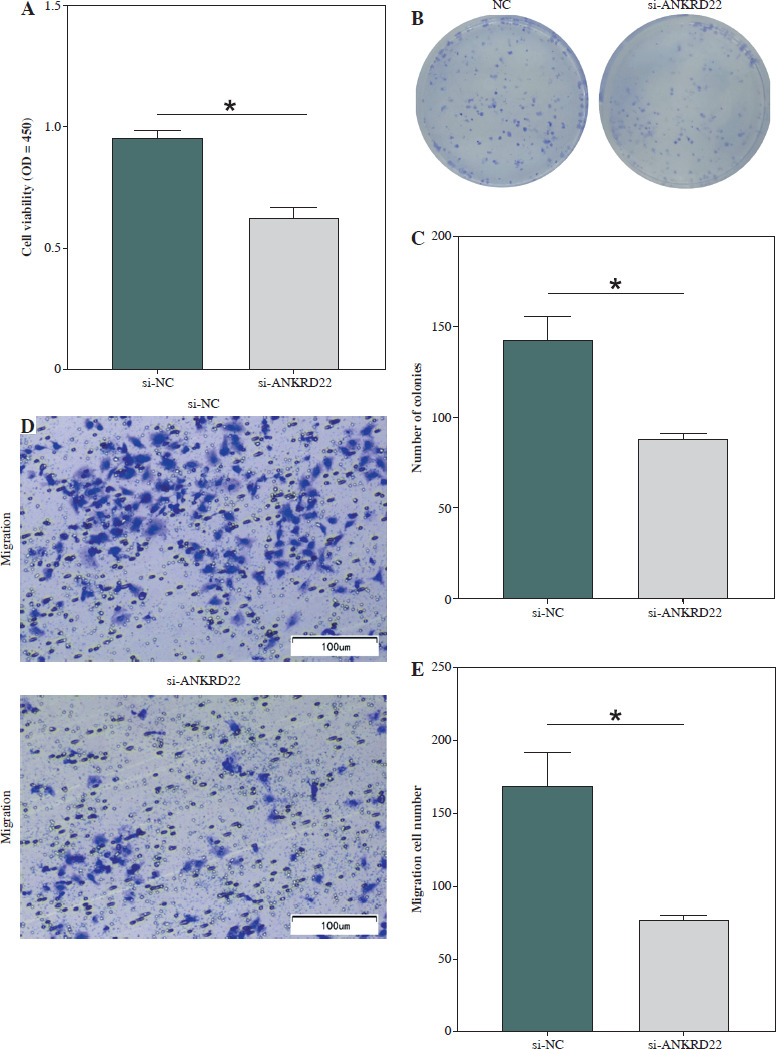
To determine the biological impact of ANKRD22 in LUAD cell lines, the effects of ANKRD22 knockdown on the neoplastic properties of A549 cells were evaluated. Utilizing the CCK-8 assay, it was observed that the knockdown of ANKRD22 led to suppression of A549 cell viability (Fig. 3A). Additionally, cell proliferation was assessed via colony formation assays, where a notable decrease in colony count was evident upon si-ANKRD22 transfection, signifying compromised cell proliferative potential (Fig. 3B, C). Furthermore, Transwell migration assays demonstrated that ANKRD22 knockdown could significantly impede the migratory ability of A549 cells (Fig. 3D, E). In summary, these outcomes implied that ANKRD22 knockdown could inhibit the malignant phenotype of LUAD cells.
Fig. 3
Reduced ANKRD22 expression suppresses malignant phenotypes in lung adenocarcinoma (LUAD). A) CCK-8 assay measured A549 cell viability. B, C) Colony formation assays assessed A549 cell proliferation. D, E) Transwell assays tested A549 cell migration. All experiments were conducted with 3 independent replicates. The data are presented as mean ± standard deviation. *p < 0.05
ANKRD22 induces macrophage M2 polarization to facilitate tumor angiogenesis in LUAD
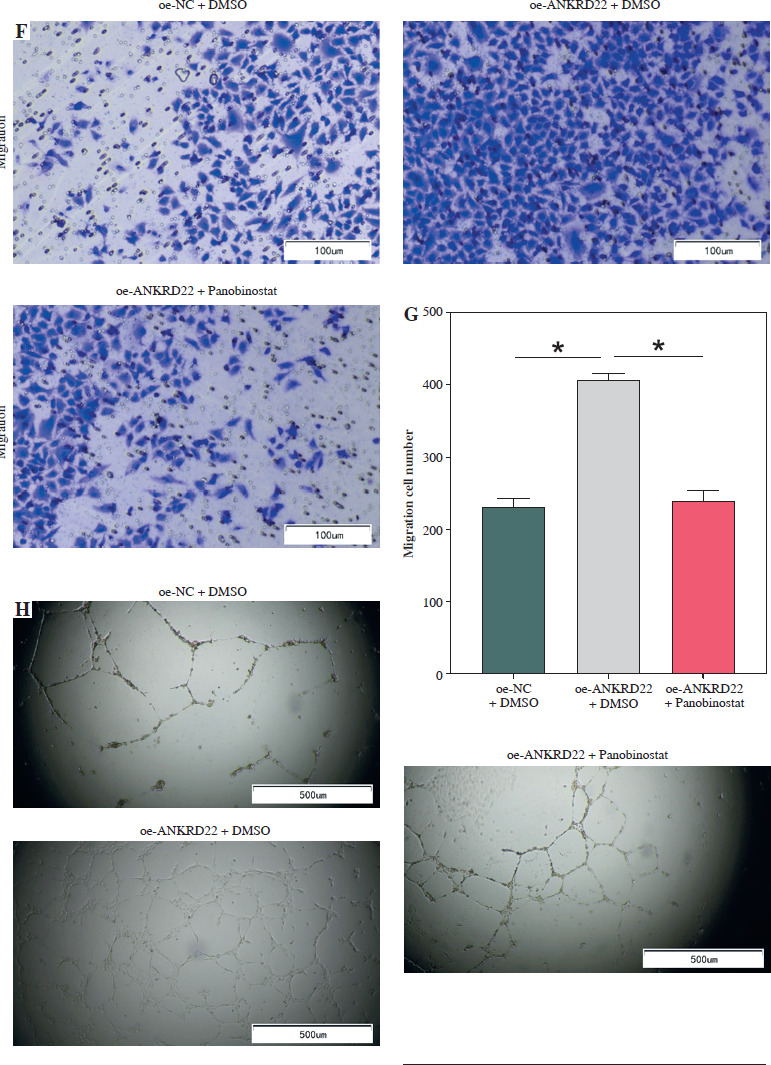
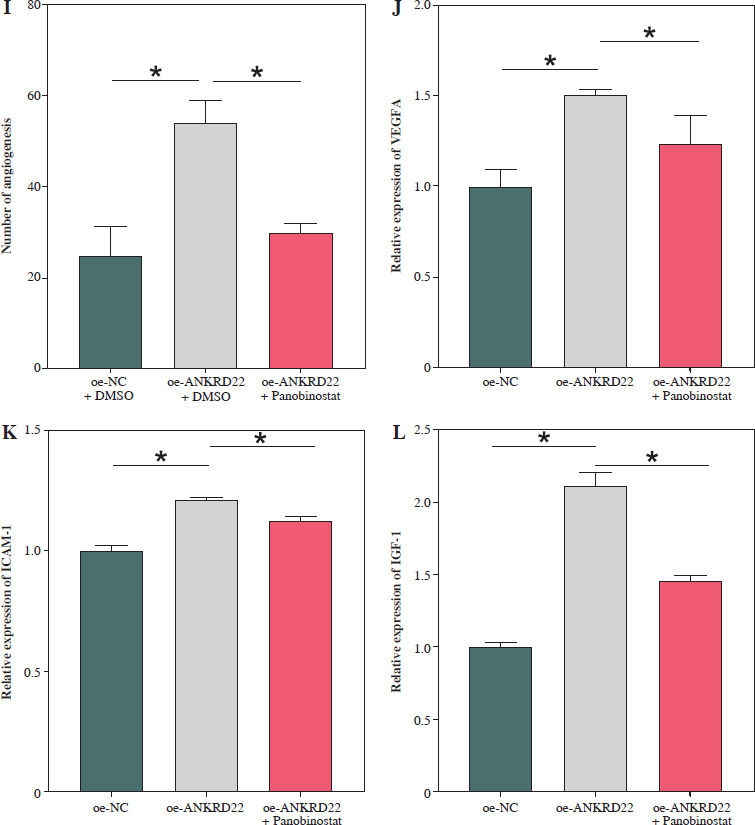
According to existing literature, macrophages within the TME are intimately associated with tumor progression, with M2 macrophages having the capacity to directly influence tumor cell survival, growth, and metastasis through the promotion of tumor angiogenesis. Given our findings, we hypothesized that alterations in the expression of the ANKRD22 gene in LUAD might impact the M2 polarization of macrophages, consequently affecting the angiogenesis within the TME. To test this hypothesis, we transfected A549 cells with the oe-ANKRD22 plasmid and introduced the macrophage M2 polarization inhibitor panobinostat to macrophages that had been educated by A549 cells to set up a rescue experiment, that is, to construct oe-NC+DMSO, oe-ANKRD22+DMSO, and oe-ANKRD22+panobinostat cell groups. qRT-PCR analyses on the three groups of macrophages showed that overexpression of ANKRD22 significantly increased mRNA levels of M2 macrophage markers IL-10 and CCL-17, which were reversed by panobinostat treatment (Fig. 4A, B). Flow cytometry was applied to detect the proportion of M2 macrophages, revealing that ANKRD22 overexpression in A549 cells enhanced M2 polarization, an effect reduced by an M2 polarization inhibitor (Fig. 4C, D). Western blot analyses on the three groups of macrophages confirmed that CD163 protein levels increased with ANKRD22 overexpression but were normalized by panobinostat (Fig. 4E). We cultured HUVEC cells with macrophage supernatants from the three groups to test the impact of M2 polarization on tumor migration and angiogenesis. Transwell assays were performed to gauge the migration ability of HUVEC cells in the three groups, showing that ANKRD22 over- expression boosted HUVEC migration, with M2 polarization further enhancing this effect (Fig. 4F, G). Angiogenesis assays revealed more vascular structures in the oe-ANKRD22 + DMSO group than in the oe-NC + DMSO group, while those in the oe-ANKRD22 + panobinostat group were markedly fewer than the oe-ANKRD22 + DMSO group (Fig. 4H, I). qRT-PCR of the mRNA expression of angiogenesis-related factors VEGFA, ICAM-1, and IGF-1 confirmed these trends (Fig. 4J-L). These results suggested that ANKRD22 may drive tumor angiogenesis in LUAD by promoting M2 macrophage polarization.
Fig. 4
ANKRD22 mediates tumor angiogenesis via M2 macrophage polarization in lung adenocarcinoma (LUAD). A, B) qRT-PCR for IL-10 and CCL-17 mRNA in three macrophage groups. C, D) Flow cytometry for M2 polarization levels (CD68+CD206+ percentage) in the three groups. E) WB for CD163 protein levels in macrophages from the three groups. The data are presented as mean ± standard deviation. *p < 0.05 F, G) Transwell assays for HUVEC cell migratory ability in the three groups. H) Tube formation tests for HUVEC cells in the three groups. The data are presented as mean ± standard deviation. *p < 0.05 I) Tube formation tests for HUVEC cells in the three groups. J-L) qRT-PCR of expression levels of angiogenic factors VEGFA, ICAM-1, and IGF-1 mRNA in HUVEC cells. All experiments were conducted with 3 independent replicates. The data are presented as mean ± standard deviation. *p < 0.05
Discussion
Through the integration of bioinformatics analysis with in vitro cellular experimentation, our study revealed elevated expression levels of ANKRD22 within LUAD tissues and cells. Subsequent functional cellular studies indicated that the overexpression of ANKRD22 was linked to the promotion of tumor angiogenesis and the M2 polarization of macrophages (derived from THP-1 cells) within the context of LUAD. Utilizing a macrophage M2 polarization inhibitor in our functional rescue experiments, we found that ANKRD22 facilitated tumor angiogenesis in LUAD by inducing M2 polarization of macrophages. These findings suggested that ANKRD22 could be a potential therapeutic target for modulating macrophage M2 polarization and advancing LUAD treatment strategies.
Current oncological literature underscores the correlation between the heightened expression of ANKRD22 and the malignant progression of various cancer types. In glial neoplasms, for example, ANKRD22 overexpression has been identified as a pivotal oncogenic factor through its positive regulation of the E2F1/MELK signaling cascade. The targeted suppression of ANKRD22 has been observed to attenuate the malignant behaviors of neoplastic cells and inhibit EMT [20]. The oncogenic influence of ANKRD22 is also observed in non-small cell lung carcinoma, implicated in the promotion of tumor cell proliferation via the upregulation of E2F1 and augmentation of cell cycle progression [17]. Our study echoed these findings, showing that silencing ANKRD22 notably reduces the proliferative and migratory abilities of LUAD cells. Researchers from the Second Affiliated Hospital of Zhejiang University School of Medicine have demonstrated that the inhibition of ANKRD22 is instrumental in the accelerated proliferation of Lgr5+ gastric epithelial progenitor cells, consequently ameliorating the inflammatory sequelae of gastric mucosal lesions [21]. Their discovery indicates that ANKRD22 may regulate immunological responses. Pan et al. [22] characterized ANKRD22 as a mitochondrial protein modulated by the TME, which is expressed in normal human gastric epithelium and shows elevated expression in activated macrophages. Thus, ANKRD22 is believed to regulate macrophage polarization.
Macrophages are predominantly derived from bone marrow-derived monocytes that migrate to the tumor periphery in response to multiple chemotactic signals. Upon entering tissues, these monocytes transform into M0 macrophages [23]. TAMs constitute an integral part of the TME and closely correlate with the onset and development of a range of tumors. The prevailing view is that TAMs can diverge into two subtypes based on the stimuli they receive from the TME. The M1 subtype typically fosters inflammation and hinders tumor progression, whereas the M2 subtype may assist in tumor immune evasion and metastasis [24]. Xu et al. [25] developed a novel prognostic model based on M2 macrophage-associated genes, discovering that TRAF2 is overexpressed in clear cell renal carcinoma and correlates with unfavorable outcomes and that TRAF2 facilitates tumor cell migration and angiogenesis by inducing macrophage M2 polarization. However, macrophage polarization is not a static process, and the M1 and M2 phenotypes are not strictly dichotomous. They are not merely mutually exclusive but often coexist, and, under specific conditions, can transform into one another. This dynamic interplay enables macrophages to sustain tissue equilibrium and systemic balance amidst fluctuating microenvironments [26]. It has been shown that the inhibition of the EGFR signaling pathway in colon cancer cells suppresses M2 polarization and enhances the M1 polarization of macrophages. On a mechanistic level, the inhibition of EGFR alters the secretion of cytokines, such as IGF-1, thereby disrupting the M1-M2 polarization of macrophages [27]. Within this study, we found that culturing with LUAD-conditioned medium that overexpresses ANKRD22 resulted in enhanced mRNA levels of the M2 macrophage markers IL-10 and CCL-17, and the protein level of CD163. There was also an increase in the ratio of CD68+CD206+ macrophages and significant upregulation of the relative mRNA expression of VEGFA, ICAM-1, and IGF-1, all of which are related to angiogenesis. Our results posit ANKRD22 as a potential target for anti-angiogenic treatments in LUAD, echoing the research by Zhang et al. [27]. However, we have not yet explored further into the molecular mechanisms by which ANKRD22 may promote M2 polarization of macrophages in LUAD.
In brief, our findings show that ANKRD22 is upregulated in LUAD tissues and cells and has the biological function of mediating macrophage polarization. That is, ANKRD22 could induce M2 polarization of macrophages in LUAD, which promotes tumor angiogenesis and hastens the malignant progression of this disease. There is a study limitation in that we did not design in vivo experiments to verify this finding. The team aims to fill this gap in our future work and probe into the mechanisms by which ANKRD22 may regulate macrophage polarization.