
Introduction
Sepsis is usually induced by an imbalance between infection and host responses [1]. Sepsis progression is a global health burden due to the associated high mortality [2]. According to previous reports, sepsis can progress to acute lung injury (ALI) in approximately 40% of cases [3]. Though great efforts have been made in the study of sepsis-induced ALI, the outcomes still remain limited. Thus, it is essential to discover effective strategies against septic lung injury.
Pyroptosis, a subtype of programmed cell death, plays an essential role in inflammation [4]. It is primarily triggered by inflammasomes and executed by downstream protease caspase-1 activation. Activated caspase-1 cleaved the substrate protein gasdermin D (GSDMD) to release the membrane perforating activity of its N-terminal domain [5]. Moreover, caspase-1 mediates the maturation and release of interleukin (IL)-1β and IL-18. Then, the mature IL-1β and IL-18 are secreted through the molecular pore formed by GSDMD on the cell membrane, and they play a pro-inflammatory physiological role [6]. In addition, NLRP1 can promote pyroptosis through the mediation of inflammatory factors (IL-18, etc.) and caspase-1 [7]. It has been illustrated that pyroptosis can rupture the plasma membrane and lead to the release of inflammatory factors in sepsis [8, 9]. Thus, pyroptosis can act as a crucial regulator in sepsis. Therefore, exploring novel pyroptosis alleviation strategies in septic lung injury is necessary.
Long non-coding RNAs (lncRNAs) are a class of non- coding RNA transcripts that are about 200 nucleotides long [10], which are vital targets in various diseases [11, 12]. In addition, lncRNAs act as vital modulators in the progression of septic lung injury. For instance, CDKN2B-AS1 could interact with LIN28B to attenuate sepsis-induced ALI via HIF-1α/NLRP3 pathway activation [13]. A recent report indicated that lncRNA NEAT1 regulated LPS-induced pyroptosis through ROCK1 mediation [14]. Meanwhile, SNHG16 is involved in multiple pulmonary diseases (pneumonia, lung cancer, etc.) [15, 16]. Nevertheless, the function of SNHG16 in sepsis-induced ALI needs to be further explored.
MicroRNAs (miRNAs) can modulate mRNA expression via mRNA degradation [17, 18]. MiRNAs are crucial mediators in various diseases, including sepsis [19, 20]. MiR-339-5p often exerts a vital function in lung cancer and sepsis-induced kidney injury [21, 22]. Nevertheless, the detailed function of miR-339-5p in sepsis-mediated ALI remains unclear. LncRNAs can inhibit miRNA activity and mediate mRNA levels as ceRNAs [23]. Database predictions indicated that SNHG16 and miR-339-5p, as well as miR-339-5p and NLR family pyrin domain containing 1 (NLRP1), have mutual binding sites. The above backgrounds suggest that SNHG16 might regulate NLRP1 by targeting miR-339-5p, thereby affecting pyroptosis in septic ALI.
Thus, it can be hypothesized that SNHG16 regulates ALI progression induced by sepsis through miR-339-5p/NLRP1 axis mediation. We hope the present study will provide a new therapeutic target for septic ALI treatment.
Material and methods
Cell culture
Human bronchial epithelial cells (BEAS-2B) were purchased from the American Type Culture Collection (Manassas, VA, USA). They were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% FBS (Thermo Fisher Scientific) at 37°C and 5% CO2. To mimic sepsis-induced ALI in vitro, cells were supplemented with LPS (0.1, 1, and 10 µg/ml, Sigma, St. Louis, MO, USA) for 24 h [24].
Cell transfection
BEAS-2B cells (3 × 105 per well) were transfected with SNHG16 small-interfering RNAs (si-SNHG16), negative control siRNA (si-NC), pcDNA3.1 (oe-NC), or pcDNA3.1-NLRP1 (oe-NLRP1). For miR-339-5p overexpression/downregulation, BEAS-2B cells were transfected with miR-339-5p mimics/inhibitor. All the vectors were obtained from GenePharma (Shanghai, China) and transfected into cells using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s protocol [25].
RT-qPCR
Total RNA was extracted from cell lines (3 × 105 per well) using TRIzol (TaKaRa, Tokyo, Japan). To detect mRNA levels, cDNA was synthesized using the PrimeScript RT kit (ELK biosciences, Wuhan, China). For miRNAs, cDNA was synthesized using the First Strand cDNA Synthesis Kit (ELK biosciences). Subsequently, RT-qPCR was performed using the SYBR premix Ex Taq II kit (Takara). Real-time qPCRs were performed in triplicate under the following protocol: 2 min at 94°C, followed by 35 cycles (30 s at 94°C and 45 s at 55°C). For mRNA, GAPDH was selected as a normalized control gene, and U6 was selected as the internal control of miRNA. The sequences for primers were as follows: SNHG16 F, 5'-CCCAAGCTTGCGTTCTTTTCGAGGTCGGC-3' and R, 5'-CCGGAATT CTGACGGTAGTTTCCCAAGTT-3'; NLRP1 F, 5'-GCAGTGCTAATGCCCTGGAT-3' and R, 5'-GAGCTTGGTAGAGGAGTGAGG-3'; GAPDH F, 5'-GAGTCCACTGGCGTCTTCA-3' and R, 5'-GGTCATGAGTCCTTCCACGA-3'; U6: F, 5'-CTCGCTTCGGCAGCACAT-3' and R, 5'-AACGCTTCACGAATTTGCGT-3'. Data were quantified using the 2-ΔΔt method.
Western blotting detection
Total protein was isolated from cells using the RIPA buffer (Beyotime, Shanghai, China). Proteins were quantified with the BCA kit (Beyotime). Subsequently, proteins (40 µg per lane) were separated using SDS-PAGE gel (10%) and then transferred to PVDF membranes (Beyotime). The membranes were incubated with primary antibodies overnight at 4°C after being blocked with 10% skim milk for 1 h. After that, membranes were incubated with HRP-conjugated secondary antibodies (ab6721, 1 : 5000, Abcam, Cambridge, MA, USA) for 1 h. Finally, membranes were scanned using the Odyssey Imaging System. The antibodies were as follows: anti-NLRP1 (Abcam, ab36852, 1 : 1000), anti-ASC (Abcam, ab283684, 1 : 1000), anti-cleaved caspase-1 (CST, MA, USA, 4199S, 1 : 1000), anti-cleaved gasdermin D (GSDMD; CST, 36425S, 1 : 1000), and anti-GAPDH (CST, 5174, 1 : 1000).
Cell Counting Kit-8 assay
BEAS-2B cells (5 × 103 per well) were cultured overnight. After 48 h of treatment, cells were treated with Cell Counting Kit-8 (CCK-8) reagents (10 µl, C0037, Beyotime) and further incubated at 37°C for 2 h. Finally, cell absorbance was measured using a microplate reader (450 nm, Invitrogen).
Lactate dehydrogenase detection
Cytotoxicity was measured using the lactate dehydrogenase (LDH) kit (C0017; Beyotime). Cells (5 × 103/well) were seeded into 96-well cell culture plates. Then, the relevant operations were carried out according to the instructions of the kit. LDH levels were measured using a microreader at 490 nm (Invitrogen).
ELISA
The supernatants of BEAS-2B cells (5 × 103 cells per well) were collected via centrifugation and then used to investigate inflammatory factors. The levels of interleukin (IL)-18 (RAB0543, Sigma) and IL-1β (900-K95, Thermo Fisher Scientific) were investigated using ELISA kits. Absorbance (450 nm) was detected using a microreader (Invitrogen).
Cell pyroptosis detection
Pyroptosis was detected using the FAM-FLICA Caspase-1 Kit (Sigma). In brief, cells were stained with the caspase-1 probe for 1 h at 37oC and then stained with PI for 10 min. Thereafter, the cells were analyzed using a flow cytometer (Becton, Dickinson and Company, Franklin Lake, NJ, USA). The percentage of caspase-1+ and PI+ cells was calculated as the pyroptosis rate.
Dual luciferase reporter assay
Wild type (WT)/mutant (MUT) of SNHG16 and 3'-UTR of NLRP1 mRNA sequences containing miR-339-5p binding sites were synthesized from GenePharma, after which they were cloned into pmirGLO vectors. The SNHG16 (WT/MUT) or NLRP1 (WT/MUT) recombinant vector was transfected into cells together with miR-339-5p mimics, miR-339-5p inhibitor, or their corresponding negative control (NC) vectors. The Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) was used to analyze luciferase activity.
Statistical analysis
All the results are presented as the mean ± standard deviation (n = 3), which were three separate experiments performed in triplicate. Student’s t-test (only two groups) or the one-way analysis of variance followed by Tukey’s test (more than two groups) was used for comparisons. P < 0.05 was considered to indicate a significant difference.
Results
SNHG16 downregulation attenuated LPS-induced pyroptosis
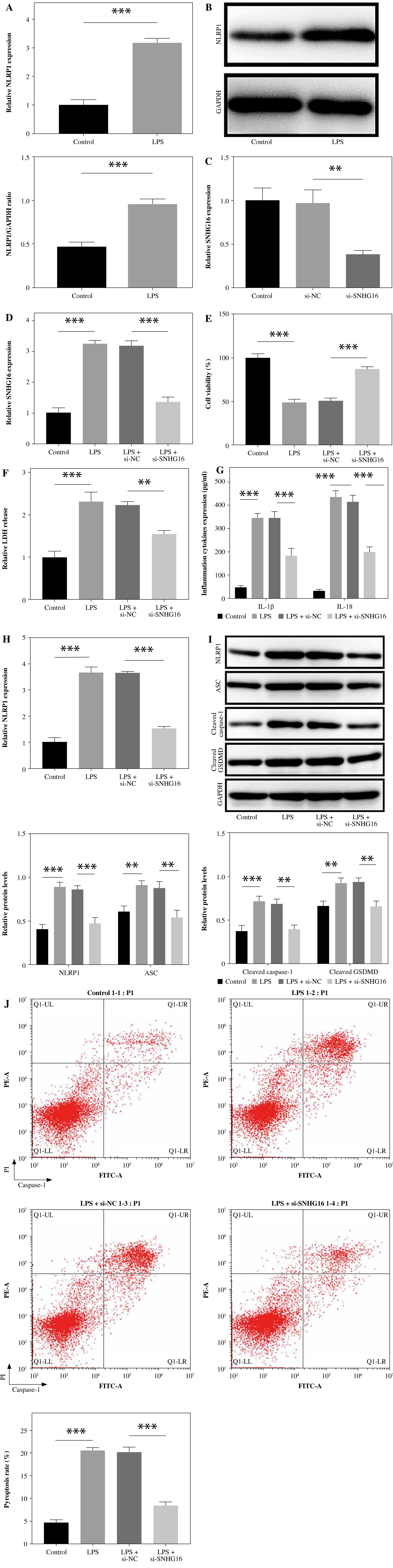
To mimic septic ALI in vitro, cells were treated with 0.1, 1, or 10 µg/ml LPS. As shown in Figure S1, LPS could inhibit cell viability and increase IL-1β, IL-18, and SNHG16 levels in a dose-dependent manner. In addition, 1 µg/ml LPS had a significant effect on the levels of IL-1β, IL-18, and SNHG16, and it reduced cell viability by about 50%. Consistently, the level of NLRP1 was increased by 1 µg/ml LPS (Fig. 1A, B). To investigate the function of SNHG16 in septic ALI, cells were transfected with SNHG16 siRNA. As expected, the SNHG16 level in BEAS-2B cells was decreased by SNHG16 siRNA (Fig. 1C). LPS-upregulated SNHG16 was also decreased by SNHG16 silencing (Fig. 1D). LPS greatly inhibited cell viability and upregulated the level of LDH, which was significantly reversed by SNHG16 siRNA (Fig. 1E, F). Meanwhile, LPS greatly induced the secretion of IL-1β and IL-18, while this phenomenon was abolished after SNHG16 knockdown (Fig. 1G). Moreover, LPS-induced NLRP1 upregulation was inhibited after SNHG16 siRNA treatment (Fig. 1H, I). LPS significantly upregulated the expression of pyroptotic proteins (ASC, cleaved caspase-1, and cleaved GSDMD) and induced BEAS-2B cell pyroptosis; however, the pro-pyroptotic effect of LPS was significantly attenuated by SNHG16 downregulation (Fig. 1I, J). In summary, SNHG16 knockdown inhibited NLRP1 expression and attenuated the pyroptotic effect of LPS.
Fig. 1
SNHG16 downregulation relieved LPS-induced pyroptosis. BEAS-2B cells were treated with LPS (1 μg/ml) for 24 h. A) The level of NLRP1 in BEAS-2B cells was detected by RT-qPCR. B) NLRP1 expression in BEAS-2B cells was analyzed via western blotting. C) Cells were transfected with sh-NC or sh-SNHG16. SNHG16 levels in BEAS-2B cells were determined by RT-qPCR. BEAS-2B cells were treated with LPS, LPS + si-NC, or LPS + si-SNHG16. D) SNHG16 levels in BEAS-2B cells were evaluated via RT-qPCR. E) BEAS-2B cell viability was tested through CCK-8 assay. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. F) LDH release was examined via the LDH kit. G) IL-18 and IL-1β levels in supernatants of BEAS-2B cells were examined via ELISA. H) The mRNA level of NLRP1 was determined by RT-qPCR. I) Protein levels of NLRP1, ASC, cleaved caspase-1, and cleaved GSDMD in BEAS-2B cells were analyzed by western blotting. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. J) BEAS-2B cell pyroptosis was tested by flow cytometry. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001
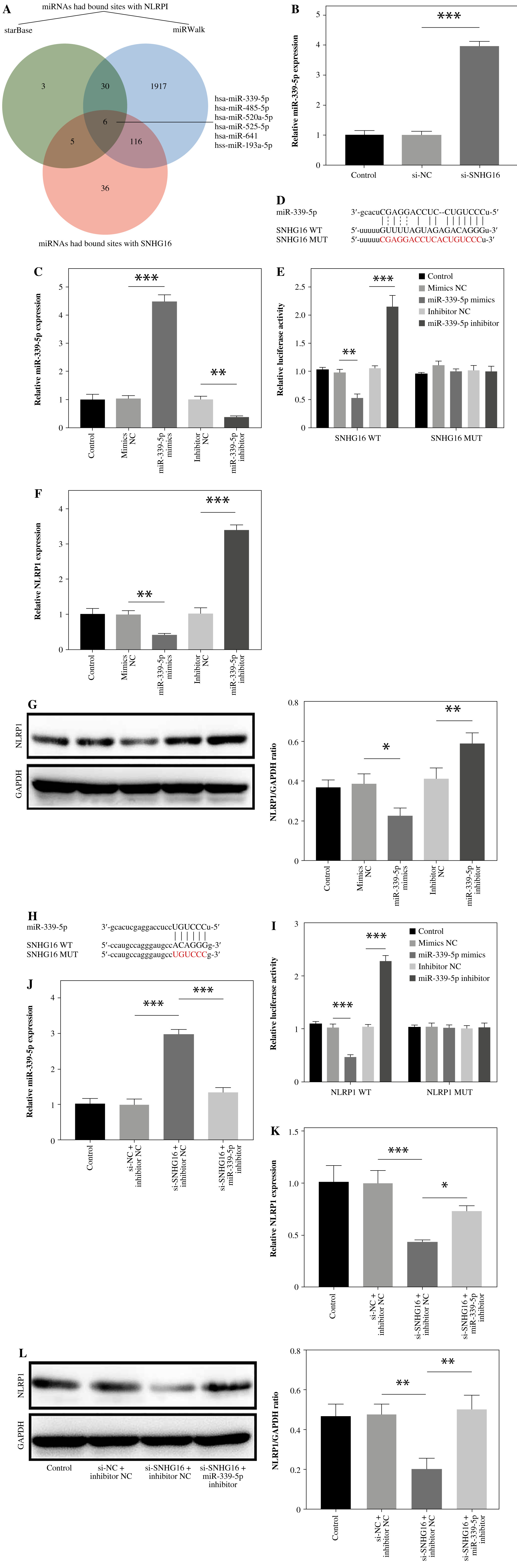
SNHG16 positively regulated NLRP1 expression through miR-339-5p sponging
Since lncRNAs have been widely reported to regulate the expression of downstream genes through the ceRNA mechanism, bioinformatics databases were used to predict miRNAs with mutually binding sites to both SNHG16 and NLRP1. As shown in Figure 2A, 6 miRNAs bound with SNHG16 and NLRP1. Importantly, the miR-339-5p level was downregulated the most by LPS in our pre-experiment (Fig. S2). Since the changes of SNHG16, NLRP1, and miR-339-5p levels in LPS-treated BEAS-2B cells were consistent with the ceRNA mechanism, miR-339-5p was selected as the major miRNA in our research. In addition, SNHG16 knockdown significantly increased miR-339-5p expression (Fig. 2B). Meanwhile, the data revealed that the miR-339-5p level in BEAS-2B cells was significantly increased by miR-339-5p mimics but downregulated by miR-339-5p inhibition (Fig. 2C). Then, we constructed SNHG16 wild and mutant type recombinant plasmids containing miR-339-5p binding sites (Fig. 2D). The result showed that miR-339-5p mimics negatively regulated the relative luciferase activity in SNHG16 WT, and miR-339-5p inhibitors exerted the opposite effect, which was not observed in the SNHG16-MUT group (Fig. 2E). The above result indicated that SNHG16 and miR-339-5p had a mutual binding relationship. Additionally, miR-339-5p could negatively regulate NLRP1 expression (Fig. 2F, G). We then predicted the binding sites of miR-339-5p and NLRP1 mRNA using the starBase database (Fig. 2H). Moreover, we verified the binding relationship between miR-339-5p and NLRP1 mRNA using the double luciferase reporter gene assay (Fig. 2I). Furthermore, SNHG16 knockdown greatly upregulated the miR-339-5p level, which was abolished by the miR-339-5p inhibitor (Fig. 2J). Concurrently, the NLRP1 level was significantly inhibited by SNHG16 silencing, while the effect of si-SNHG16 was reduced by miR-339-5p downregulation (Fig. 2K, L). To sum up, SNHG16 increased NLRP1 expression in BEAS-2B cells by binding with miR-339-5p.
Fig. 2
SNHG16 positively regulated NLRP1 expression through miR-339-5p sponging. A) The starBase and miRWalk databases were used to explore miRNAs that shared common binding sites with SNHG16 and NLRP1. B) The expression of miR-339-5p in BEAS-2B cells was investigated by RT-qPCR after si-NC or si-SNHG16 transfection. BEAS-2B cells were transfected with NC mimics, miR-339-5p mimics, inhibitor NC, or miR-339-5p inhibitor. C) The expression of miR-339-5p in BEAS- 2B cells was examined via RT-qPCR. D) The binding sites among SNHG16 and miR-339-5p were tested by the starBase database. E) The dual luciferase assay was performed to investigate luciferase activity. F) NLRP1 expression in BEAS-2B cells was examined via RT-qPCR and western blotting. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001 G) NLRP1 expression in BEAS-2B cells was examined via RT-qPCR and western blotting. H) The binding sites between NLRP1 and miR-339-5p were predicted by the starBase database. I) The dual luciferase assay was performed to measure luciferase activity. Cells were treated with inhibitor NC + si-NC, si-SNHG16 + NC inhibitor, or si-SNHG16 + miR-339-5p inhibitor. J, K) Levels of miR-339-5p and NLRP1 were examined via RT-qPCR. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. L) NLRP1 expression in BEAS-2B cells was tested by western blotting. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001
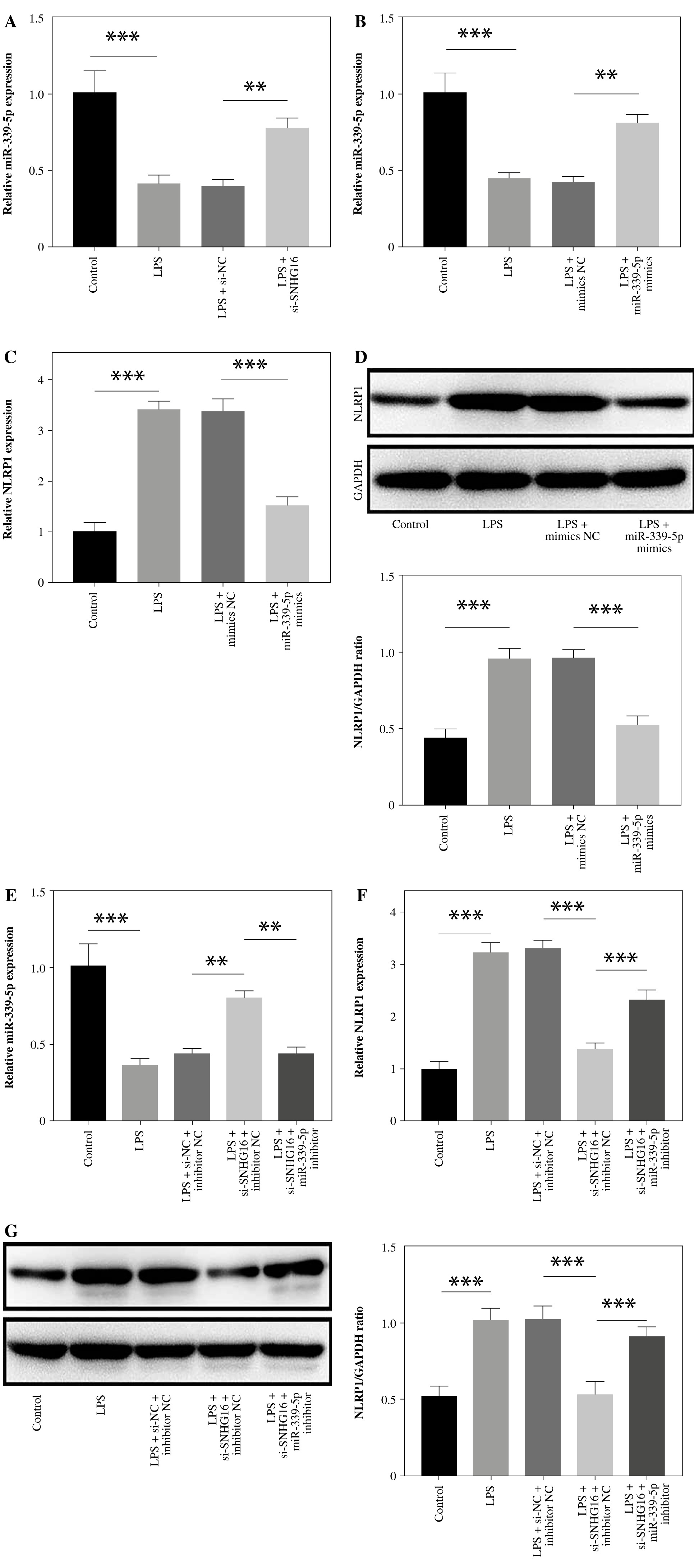
MiR-339-5p downregulation reversed the inhibitory effect of SNHG16 silencing on the NLRP1 level in LPS-treated BEAS-2B cells
To verify the relationship between SNHG16 and miR-339-5p, cells were transfected with si-SNHG16 or miR-339-5p mimics. As shown in Figure 3A, B, LPS-induced miR-339-5p downregulation in BEAS-2B cells was reversed by SNHG16 silencing or miR-339-5p mimics. In contrast, LPS-upregulated NLRP1 was significantly attenuated by miR-339-5p mimics (Fig. 3C, D). SNHG16 siRNA-mediated regulation of miR-339-5p and NLRP1 was significantly abolished by miR-339-5p inhibitor (Fig. 3E-G). Taken together, SNHG16 increased the level of NLRP1 through miR-339-5p downregulation in LPS- induced BEAS-2B cells.
Fig. 3
MiR-339-5p downregulation reversed the inhibitory effect of SNHG16 silencing on the NLRP1 level in LPS-treated BEAS-2B cells. A) BEAS-2B cells were treated with LPS, LPS + si-NC, or LPS + si-SNHG16. The level of miR-339-5p in BEAS-2B cells was determined via RT-qPCR. BEAS-2B cells were treated with LPS, LPS + mimics NC, or LPS + miR-339-5p mimics. B, C) Levels of miR-339-5p and NLRP1 were determined via RT-qPCR. D) The protein level of NLRP1 was tested by western blotting. BEAS-2B cells were treated with LPS, LPS + inhibitor NC + si-NC, LPS + si-SNHG16 + inhibitor NC, or LPS + si-SNHG16 + miR-339-5p inhibitor. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. E, F) Levels of miR-339-5p and NLRP1 in BEAS-2B cells were examined via RT-qPCR. G) NLRP1 expression in BEAS-2B cells was tested by western blotting. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001
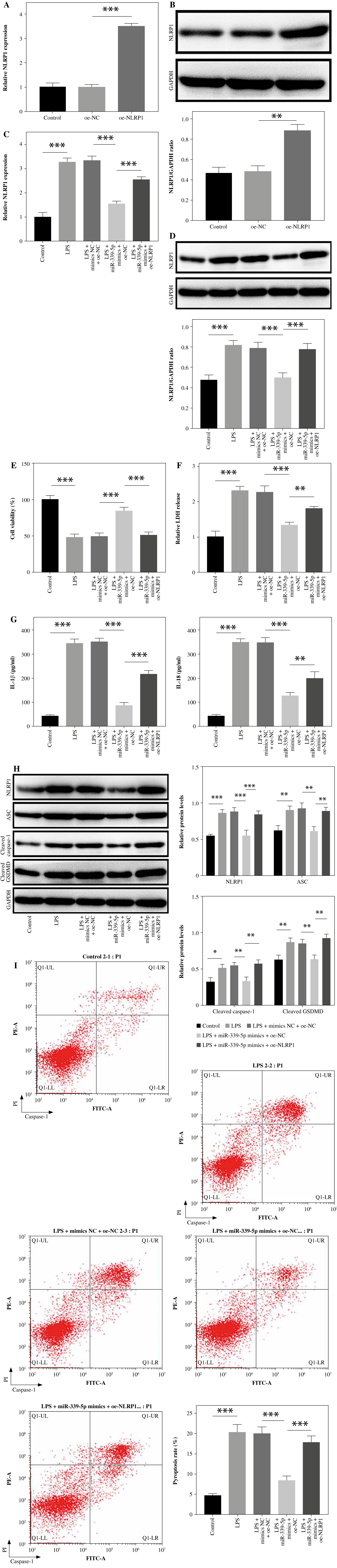
NLRP1 reversed miR-339-5p overexpression-mediated pyroptosis inhibition in LPS-induced BEAS-2B cells
Next, to investigate the function of miR-339-5p in sepsis-induced ALI, BEAS-2B cells were transfected with pcDNA3.1 NLRP1 (oe-NLRP1). As demonstrated in Figure 4A, B, oe-NLRP1 significantly increased the level of NLRP1 in BEAS-2B cells, indicating that oe-NLRP1 was successfully transfected in cells. LPS-induced NLRP1 activation was significantly relieved by miR-339-5p mimics, which was reversed by NLRP1 overexpression (Fig. 4C, D). MiR-339-5p mimics significantly reversed the effect of LPS on the viability and LDH level of BEAS-2B cells, which was significantly abolished by NLRP1 upregulation (Fig. 4E, F). Consistently, the inhibitory effect of miR-339-5p mimics on the secretion of IL-18 and IL-1β was greatly offset after NLRP1 upregulation (Fig. 4G). Additionally, NLRP1 overexpression greatly reversed the anti-pyroptotic effect of miR-339-5p mimics on LPS-treated cells through facilitating ASC, cleaved caspase-1, and cleaved GSDMD protein levels and caspase-1 activity (Fig. 4H, I). In summary, miR-339-5p ameliorated LPS-induced pyroptosis in BEAS-2B cells via NLRP1 downregulation.
Fig. 4
NLRP1 reversed miR-339-5p overexpression-mediated pyroptosis inhibition in LPS-induced BEAS-2B cells. A, B) The overexpression effect of NLRP1 was tested by RT-qPCR and western blotting after transfection with oe-NLRP1. BEAS-2B cells were exposed to LPS, LPS + mimics NC + pcDNA3.1, LPS + miR-339-5p mimics + pcDNA3.1, or LPS + miR-339-5p mimics + pcDNA3.1-NLRP1. C, D) NLRP1 levels in BEAS-2B cells were tested by RT-qPCR and western blotting. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. E) BEAS-2B cell viability was assessed by the CCK-8 assay. F) LDH release was tested using the LDH kit. G) IL-18 and IL-1β levels in cell supernatants were tested by ELISA. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. H) Protein levels of ASC, cleaved caspase-1, and cleaved GSDMD in BEAS-2B cells were detected by western blotting. I) BEAS-2B cell pyroptosis was determined by flow cytometry. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. I) BEAS-2B cell pyroptosis was determined by flow cytometry. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001
NLRP1 upregulation abolished SNHG16 knockdown-reduced pyroptosis
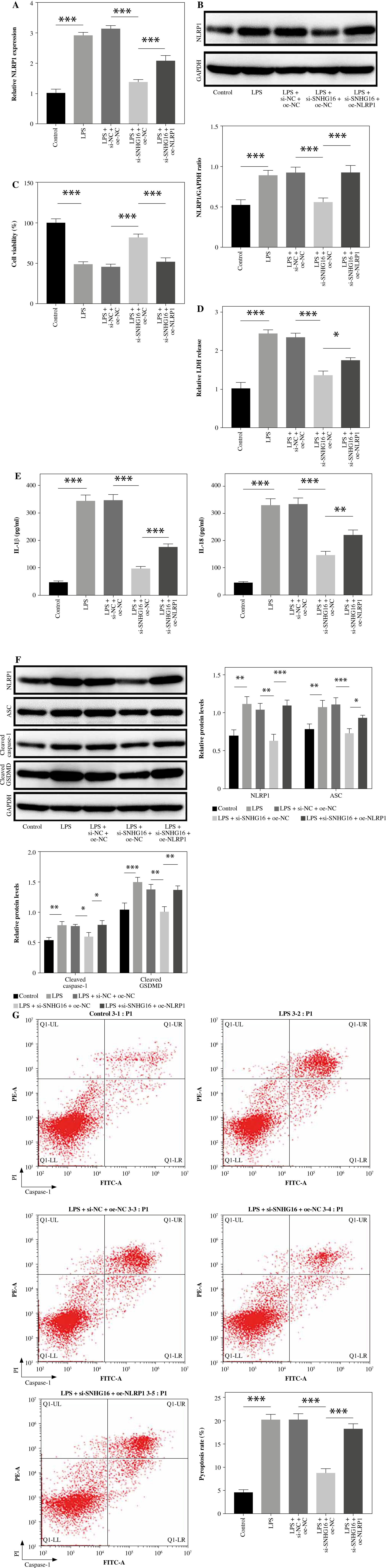
To further confirm the relationship between SNHG16 and NLRP1 in septic ALI, cells were co-transfected with SNHG16 siRNA and oe-NLRP1. As indicated in Fig- ure 5A, B, si-SNHG16-induced NLRP1 downregulation was significantly rescued in the presence of oe-NLRP1. In addition, the inhibitory effect of si-SNHG16 on LPS-induced cell injury was greatly abolished by NLRP1 upregulation (Fig. 5C, D). Consistently, NLRP1 overexpression reversed SNHG16 siRNA-inhibited inflammation in LPS-treated BEAS-2B cells (Fig. 5E). Furthermore, NLRP1 overexpression significantly abolished the anti-pyroptotic effect of si-SNHG16 on LPS-treated BEAS-2B cells (Fig. 5F, G). Collectively, SNHG16 aggravated LPS-induced BEAS-2B pyroptosis through NLRP1 upregulation.
Fig. 5
NLRP1 upregulation abolished SNHG16 knockdown-reduced pyroptosis. BEAS2B cells were transfected with LPS, si-NC + LPS + pcDNA3.1, LPS + si-SNHG16 + pcDNA3.1, or LPS + si-SNHG16 + pcDNA3.1-NLRP1. A, B) NLRP1 levels in BEAS-2B cells were tested by RT-qPCR and western blotting. C) BEAS-2B cell viability was assessed by the CCK-8 assay. D) The LDH kit was used to test for LDH release. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. E) IL-1β and IL-18 levels in supernatants of BEAS-2B cells were tested by ELISA. F) Protein levels of ASC, cleaved caspase-1, and cleaved GSDMD in BEAS-2B cells were detected by western blotting. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. G) BEAS-2B cell pyroptosis was determined by flow cytometry. n = 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001 G) BEAS-2B cell pyroptosis was determined by flow cytometry. n = 3 per group. * p < 0.05, **p < 0.01, ***p < 0.001
Discussion
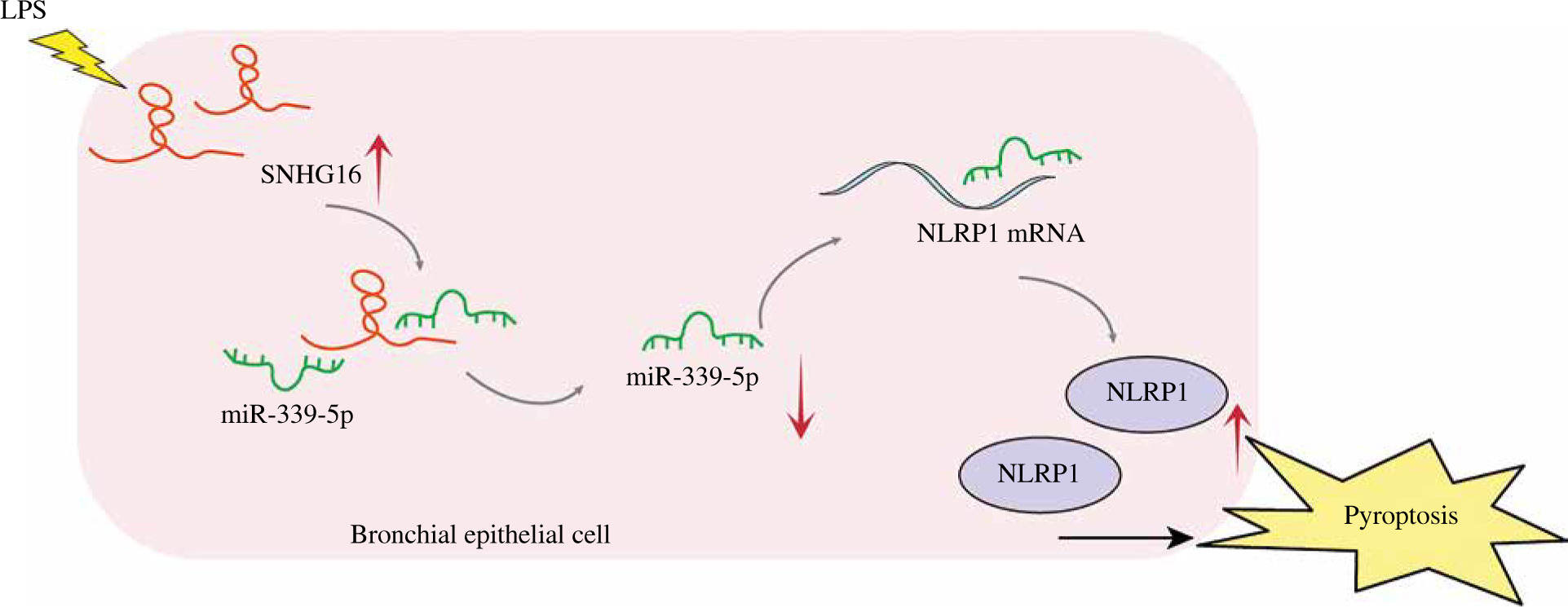
Sepsis, a condition induced by bacterial infection, is associated with high mortality rates. Without proper treatment, sepsis could cause multi-organ failure, tissue damage, and death [26, 27]. As a frequent complication of sepsis, ALI can lead to acute respiratory failure [28]. Therefore, it is essential to develop a therapeutic strategy against septic ALI. Pyroptosis can induce lung injury in sepsis. For example, Liu et al. found that the downregulation of STAT3 phosphorylation could inhibit pyroptosis to alleviate sepsis-induced ALI [29]; lncRNA NEAT1 regulated LPS-induced pyroptosis through ROCK1 mediation [14]. In this study, we found that SNHG16 facilitated LPS-induced bronchial epithelial cell pyroptosis through miR-339-5p/NLRP1 axis mediation, and this study might provide new evidence for increasing the clinical value of SNHG16 in ALI (Fig. 6).
Fig. 6
The mechanism underlying the function of SNHG16 in septic ALI. SNHG16 sponged miR-339-5p to indirectly upregulate NLRP1 in LPS-treated bronchial epithelial cells BEAS-2B. Then, the upregulated NLRP1 promoted activation of caspase-1 and secretion of pyroptosis-related factors (IL-1β and IL-18), thus aggravating BEAS-2B pyroptosis
SNHG16 plays a crucial role in the pathogenesis of inflammatory diseases. For instance, SNHG16 rescued the effects of miR-15a/16 on neonatal sepsis-induced RAW264.7 cell inflammation [30]. Also, a previous study showed that SNHG16 downregulation attenuated the development of sepsis-induced ALI through miR-128-3p/HMGB3 axis regulation [31]. Our research further explored the mechanism by which SNHG16 regulated pyroptosis in LPS-induced BEAS-2B cells. We found that SNHG16 silencing reversed the promoting effect of LPS on pyroptosis. To the best of our knowledge, this study is the first to demonstrate the function of SNHG16 in pyroptosis during the pathogenesis of septic ALI.
At present, reports of SNHG16 in the clinical study of sepsis are scarce and not in-depth enough. Zhang et al. found that a higher SNHG16 level was associated with a lower incidence of acute respiratory distress syndrome (ARDS) at the clinical level [32], which seems to differ from the findings of our study. Our findings and those of other researchers suggested that SNHG16 plays a promoting role in sepsis-induced ALI and the inflammatory response [33-35]. For example, Zhang et al. found that SNHG16 was highly expressed in acute pneumonia and LPS-induced human fibroblasts (WI-38), and SNHG16 could induce apoptosis and inflammatory injury in LPS-treated A549 cells by targeting the miR-370-3p/IGF2 axis [35]. Xia et al. suggested that SNHG16 knockdown could reverse LPS-induced apoptosis, autophagy, and inflammatory responses in WI-38 cells [34]. ALI and ARDS have similar pathophysiologic changes, and severe ALI, or the final severe stage of ALI is defined as ARDS. The current discrepancy in results among existing studies may be related to various factors, such as cell types and disease severity, which need to be further deciphered and differentiated by more studies.
MiRNAs participate in sepsis-induced ALI progression. Shen et al. reported that miR-125b-5p inhibited the ferroptosis of pulmonary microvascular endothelial cells through regulating the Keap1/Nrf2/GPX4 axis in septic lung injury [36]; circ_0001498 could aggravate LPS-induced inflammation in septic ALI by interacting with miR-574-5p [37]. Meanwhile, miR-339-5p participated in the progression of cancer and inflammatory diseases [38, 39]. Notably, miR-339-5p participated in the pathogenesis of the LPS-induced inflammatory response in RAW264.7 cells [21]. The present study demonstrated that miR-339-5p was sponged by SNHG16 and directly targeted NLRP1 mRNA. In addition, miR-339-5p inhibited LPS-induced BEAS-2B cell pyroptosis, and NLRP1 overexpression reversed this phenomenon. This study demonstrated that SNHG16 could positively regulate NLRP1 expression through interactions with miR-339-5p, thus mediating LPS-induced pyroptosis in BEAS-2B cells.
NOD-like receptor protein 1 (NLRP1), which belongs to the NOD-like receptor family, is considered a crucial inflammasome [40]. NLRP1 serves as an important modulator in pyroptosis. As evidence, NLRP1 promoted pyroptosis by binding to miR-637 during vascular endothelial injury, and it induced neuronal pyroptosis through STING/IRE1alpha pathway mediation after traumatic brain injury in mice [41, 42]. A previous study suggested that NLRP1 is an important risk factor for sepsis [43], and it could promote septic lung injury [44]. Consistently, our findings revealed that NLRP1 overexpression abolished the anti-pyroptotic effect of SNHG16 siRNA during septic ALI progression. These findings implied that SNHG16 silencing could reverse LPS-induced pyroptosis through miR-339-5p/NLRP1 axis mediation.
Nevertheless, this study had several limitations, including the following: 1) other downstream targets of SNHG16 in sepsis-induced ALI remain unexplored; 2) there were no animal studies to further assess the impact of SNHG16 in sepsis-induced ALI. Hence, more investigations are needed in the future.
In summary, SNHG16 has the potential to be a therapeutic target against septic ALI. Our study will hopefully provide a new theoretical basis for exploring new therapeutic strategies for septic ALI treatment.