
Introduction
Osteoarthritis (OA) is a chronic degenerative disease that causes pain and dysfunction in human limbs [1, 2]. Articular cartilage lesions are considered to be one of the key drivers of OA [3]. Chondrocytes are the only cell type present in articular cartilage and can regulate cartilage homeostasis by synthesizing extracellular matrix (ECM) rich in type II collagen, proteoglycans, and macromolecules [4, 5]. Chondrocyte apoptosis, elevated levels of pro-inflammatory factors and imbalance of ECM metabolism have been found to play critical roles in the occurrence and development of OA [6]. In recent years, an increasing number of studies have shown that inflammation present in OA is the main pathogenic factor for joint injury [7]. For example, metformin could slow down chondrocyte apoptosis and catabolism, and inhibit synovial macrophage infiltration and macrophage production of inflammatory factors, thus achieving relief of OA [8]. Therefore, this study will take this as an entry point to further investigate in depth the molecular mechanisms of articular cartilage damage in OA, to find out the key links that clarify the progression of OA, thus providing a new perspective for the clinical prevention and treatment of OA.
Long non-coding RNAs (lncRNAs) are noncoding RNAs longer than 200 nt that have no protein-coding function [9]. However, they can interact with protein, DNA, RNA, etc., and participate in cellular biological processes at multiple levels such as gene transcription, post-transcriptional regulation, and epigenetic regulation [10]. Many studies have shown that lncRNAs play an important role in the regulation of chondrocyte injury of OA [11-13]. Notably, Steinbusch et al. found that lncRNA, the RNA component of the mitochondrial RNA processing endoribonuclease (RMRP), as an important factor in the regulation of cartilage differentiation, was positively correlated with the chondrocyte hypertrophy phenotype [14]. In addition, previous studies have proved that mutations in RMRP caused achondroplasia [15]. RMRP mutations disrupted cartilage formation and ossification, inhibited chondrocyte proliferation and promoted apoptosis [16]. Most importantly, studies have shown that the expression of RMRP was significantly increased in cartilage tissues of OA patients, whereas knockdown of RMRP promoted the proliferation of chondrocytes and inhibited the apoptosis of chondrocytes in OA [17]. Therefore, RMRP may be a key factor in chondrocyte injury in OA.
Forkhead box protein C1 (FOXC1) is a transcription factor of the forkhead family, which has an essential function in various pathophysiological and biological processes, including metabolism, differentiation, proliferation, apoptosis, migration, and invasion [18]. Notably, recent research indicated that FOXC1 expression was significantly elevated in OA [19]. Another study also reported that miR-204-5p inhibited inflammation of synovial fibroblasts in OA by inhibiting FOXC1 [20]. In addition, Yuan et al. found that miR-138-5p targeted the expression of FOXC1 and promoted the degradation of ECM induced by interleukin (IL)-1β [21]. Interestingly, the starBase (https://starbase.sysu.edu.cn/index.php) analysis found a potential binding site between FOXC1 and RMRP. Therefore, it is not difficult to speculate that RMRP may regulate FOXC1 to promote chondrocyte apoptosis and inflammation, thereby affecting the progression of OA disease.
In recent years, the medical community has gradually recognized adipokines as a new potential breakthrough point for OA treatment [22]. Adipokines may significantly affect the occurrence or progression of OA by modulating inflammation, metabolic homeostasis, ECM remodeling, chondrocyte apoptosis and autophagy [23-26]. Notably, adipokine retinol-binding protein 4 (RBP4) was first revealed to be produced by cartilage and highly expressed in chondrocytes of OA patients [27]. Studies have demonstrated that activation of c-Jun N-terminal kinase (JNK) leads to phosphorylation of c-Jun, thereby inhibiting the production of proteoglycans, stimulating the expression of matrix metalloproteinase 13 (MMP13), and ultimately leading to cartilage destruction [28]. It was reported that RBP4 induced the release of inflammatory factors through toll-like receptor 4 and JNK signaling pathways, mediating the development of inflammatory immune responses [29]. More importantly, from the JASPAR (http://jaspar.genereg.net/) analysis, there was a binding site between FOXC1 and the RBP4 promoter region. Accordingly, FOXC1 may induce chondrocyte injury in OA by activating the JNK signaling pathway through the upregulation of RBP4.
In summary, we speculate that RMRP up-regulates the expression of RBP4 by binding to FOXC1, thereby activating the JNK pathway and ultimately promoting the damage of chondrocytes in OA. Investigating this mechanism will help to provide new insights into the clinical treatment of chondrocyte injury in OA.
Material and methods
Collection of cartilage tissues
Knee cartilage tissues were collected from 10 OA patients (55 ±5 years) and 10 amputees without a history of OA. This project was approved by the Ethics Committee of Hainan General Hospital, and the ethics number is 2022-190. All participants signed informed consent forms. Tissue samples were cut into small pieces, some of which were fixed with 4% paraformaldehyde, and some snap-frozen in liquid nitrogen and stored at –80°C for subsequent experiments.
Cell culture
Chondrocytes were isolated from the cartilage tissue of amputees without a history of OA. Specifically, knee cartilage tissues were placed in a 10 cm dish in small pieces under 1 mm3. Then, Dulbecco’s modified eagle medium (DMEM, Beyotime, Shanghai, China) containing 0.2% type II collagenase was added and digested in a 37°C, 5% CO2 incubator for 6 hours. Next, the liquid with the tissue mass was introduced into the centrifuge tube for centrifugation to remove the supernatant. Then, the cells were cultured in DMEM containing 10% fetal bovine serum (FBS, Beyotime, Shanghai, China) in an incubator under 5% CO2 at 37°C, and the medium was changed every 48 hours. After 80-90% of the cells were fused, the in vitro OA model was constructed in the third-generation cells. During the logarithmic growth phase of chondrocytes, a dose of 5 µg/ml lipopolysaccharide (LPS, Sigma, St. Louis, MO, USA) was administered for 12 hours to establish an OA cell model for subsequent experiments.
Cell transfection
Short hairpin RNAs (shRNAs) of RMRP, FOXC1 and RBP4 were synthesized by RiboBio (Guangzhou, China). In addition, the full-length genes of RMRP and FOXC1 were cloned into the lentiviral vector (Invitrogen, Carlsbad, CA, USA), and then lentiviral vectors were transfected into chondrocytes using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). Cells were collected after 48 hours of transfection for subsequent experiments.
Cell counting kit-8 (CCK-8) assay
Cell viability was determined using Cell Counting Kit-8 (Beyotime, Shanghai, China). Chondrocytes were seeded into 96-well plates (1 × 105 cells/well) (Corning, NY, USA) for 24 hours. According to the instructions of the kit, 10 µl of CCK-8 solution was added to each well and incubated at 37°C for 1 hour. Finally, absorption was measured at a wavelength of 450 nm using a SpectraMax iD3/iD5 microplate reader (Molecular Devices, San Jose, CA, USA).
Terminal deoxynucleotidyl transferase (TDT)-mediated dUTP nick end labeling (TUNEL) assay
Apoptosis of chondrocytes was determined using TUNEL staining. After chondrocytes were vaccinated into 12-well plates (Corning, NY, USA) and cultured for 24 hours, fixative (4% paraformaldehyde) was added to the cells and fixed for 30 minutes. Subsequently, the cells were stained using TUNEL reagent (Beyotime, Shanghai, China) for 55 minutes at room temperature. Nuclei were stained with 4',6-diamidino-2-phenylindole (Beyotime, Shanghai, China). Fluorescence signals were acquired with a fluorescent microscope (Leica, Wetzlar, Germany).
Enzyme-linked immunosorbent (ELISA) assay
The experimental procedure was carried out according to the instructions of tumor necrosis factor α (TNF-α), IL-1β, IL-6 and IL-8 ELISA kits (Meimian, Jiangsu, China), and then the absorbance at 450 nm was measured on a SpectraMax iD3/iD5 microplate reader (Molecular Devices, San Jose, CA, USA). The standard curves were created using Origin software (OriginLab, Northampton, MA, USA).
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted from chondrocytes or cartilage tissue using Trizol reagent (Takara, Dalian, China). First-strand cDNA was synthesized using a PrimeScript RT reagent Kit (Takara). RT-qPCR was performed to detect the expression of RMRP, RBP4 and FOXC1 using TB Green Premix Ex Taq II (Takara). Using GAPDH as an internal reference, relative mRNA levels were calculated using the 2-ΔΔCt equation. The primers used in the study were as follows (5'-3'):
RMRP-F: TAGACATTCCCCGCTTCCCACTC,
RMRP-R: AACTAGAGGGAGCTGACGGATGAC;
RBP4-F: GGTCCTATATAGAGATAAAACC,
RBP4-R: CCACTAGCTAGCAGCAAAATCGC;
FOXC1-F: TAGCTACATCGCGCTCATCA,
FOXC1-R: ACCTTGACGAAGCACTCGTT;
GAPDH-F: TGAAGGTCGGTGTGAACGGATTTGGC,
GAPDH-R: CATGTAGGCCATGAGGTCCACCAC.
Western blot
Total protein was collected by lysing chondrocytes with RIPA buffer (Beyotime, Shanghai, China). Protein concentrations were measured with a BCA assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Then proteins were isolated using 10% SDS-PAGE gels and transferred to PVDF membranes (Roche, Basel, Switzerland). After blocking with 5% skim milk powder, membranes were incubated with antibodies targeting FOXC1 (ab227977, 1 : 1000, Abcam, Cambridge, MA, USA), RBP4 (ab109193, 1 : 5000, Abcam), B cell lymphoma/leukemia-2 (Bcl-2, ab182858, 1 : 1000, Abcam), Bcl-2 associated X protein (Bax, ab32503, 1 : 2000, Abcam), caspase 3 (ab32351, 1 : 5000, Abcam), MMP13 (ab39012, 1 : 2000, Abcam), collagen II (ab34712, 1 : 2000, Abcam), aggrecan (ab3778, 1 : 1000, Abcam), JNK (PA5-105049, 1 : 1000, Thermo Fisher Scientific), p-JNK (ab307802, 1 : 1000, Abcam) and GAPDH (ab8245, 1 : 2000, Abcam) overnight at 4°C. After washing the membrane with PBST, it was incubated with IgG (ab6721, 1 : 5000, Abcam) for 2 hours at room temperature. Finally, chemiluminescent substrates (Meilunbio, Dalian, China) were employed to identify the protein bands, which were then analyzed using Image J software (GE Healthcare, Sunnyvale, CA, USA). GAPDH was chosen as a control for endogenous normalization.
RNA immunoprecipitation (RIP) assay
The binding relationship between RMRP and FOCX1 was detected using the Magna RIP Kit (Millipore, Billerica, MA, USA). Next, the chondrocytes were lysed with RIPA buffer and cell extracts were collected. They were incubated overnight with A/G magnetic beads bound to FOXC1 (ab227977, 1 : 1000, Abcam, Cambridge, MA, USA) or IgG (ab6721, 1 : 5000, Abcam) antibodies at 4°C. After digestion of the precipitation complex using protease K buffer, RNA was extracted and the relative expression level of RMRP was assessed by RT-qPCR assay.
RNA pull-down assay
After chondrocytes were lysed with RIPA buffer (Beyotime, Shanghai, China) the cell lysate was fully incubated with a negative or RMRP probe designed by RiboBio (Guangzhou, China) and treated with streptavidin magnetic beads (Life Technologies, Carlsbad, CA, USA) for an additional 1 hour. The protein bound to the RMRP probe was then eluted and analyzed using western blot.
Dual luciferase reporter assay
The predicted binding sites of FOXC1 on RBP4 (WT-RBP4) and their mutated site (MUT-RBP4) were cloned into the firefly luciferase gene in the PGL3 vector (Promega, Madison, WI, USA). Chondrocytes were cultured in 24-well plates (Corning, NY, USA) for 12 hours, then the cells were co-transfected with the WT-RBP4/MUT-RBP4 reporter gene plasmid and FOXC1-pcDNA3.1. After 48 hours, luciferase activity was evaluated using a dual luciferase reporter detection system.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed according to MAGnify Chromatin Immunoprecipitation System Kit instructions (Thermo Fisher Scientific, Waltham, MA, USA). After chondrocytes were fixed with formaldehyde, the cells were lysed with RIPA buffer. Then, cell lysates were collected and their DNA fragmented by sonication. Next, the lysates were incubated with FOXC1 (ab227977, 1 : 1000, Abcam, Cambridge, MA, USA) antibody or IgG (ab6721, 1 : 5000, Abcam) antibody overnight at 4°C. Finally, the DNA was enriched and subjected to PCR.
Statistical analysis
All experimental data were analyzed by GraphPad Prism 9.0 and expressed as mean ± standard deviation. Between-group differences and multi-group comparisons were analyzed by Student’s t test or one-way analysis of variance (ANOVA), respectively. All experiments were repeated at least 3 times. P < 0.05 was considered statistically significant.
Results
RMRP, FOXC1 and RBP4 were upregulated in OA and LPS-induced chondrocytes
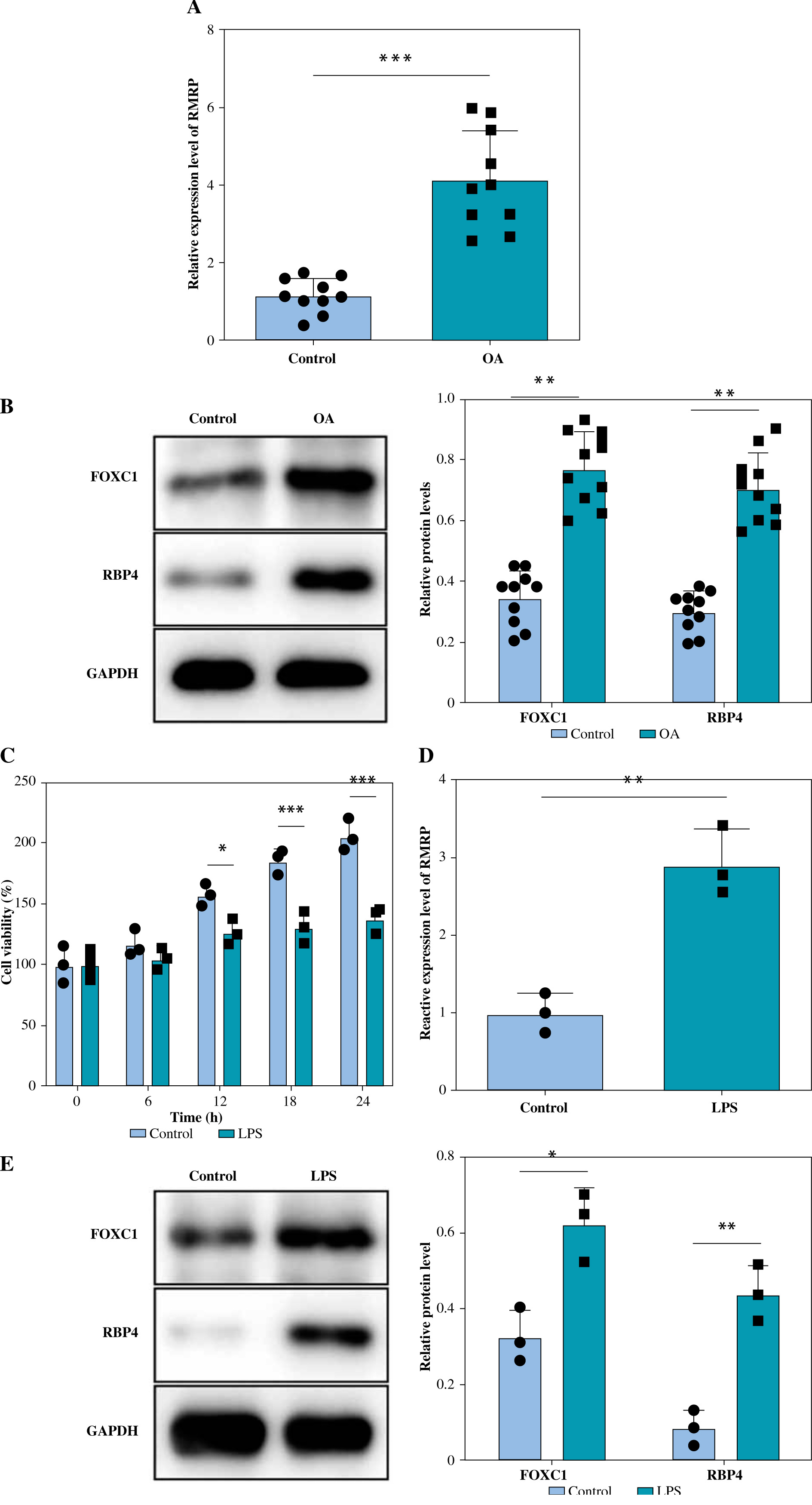
RT-qPCR was performed to detect the expression of RMRP in cartilage tissues of OA patients as well as in normal knee cartilage tissues. It was observed that the level of RMRP was significantly higher in OA patients than in the normal group (Fig. 1A). In addition, the levels of FOXC1 and RBP4 were higher in OA cartilage tissues (Fig. 1B). In vitro, LPS was added to chondrocytes to construct an OA cell model, and it was observed that the chondrocyte viability was lowered by LPS (Fig. 1C). However, the results showed that the RMRP level was elevated in LPS-induced chondrocytes (Fig. 1D). Similarly, the expression levels of FOXC1 and RBP4 were significantly increased in chondrocytes under LPS treatment (Fig. 1E). Altogether, the above results demonstrated that the levels of RMRP, FOXC1 and RBP4 were increased in OA and LPS-induced chondrocytes.
Fig. 1
RMRP, FOXC1 and RBP4 were upregulated in osteoarthritis (OA) and LPS-induced chondrocytes. Knee cartilage tissues (human) were collected from patients with OA (n = 10) and amputees without a history of OA (n = 10). A) RT-qPCR was used to detect the level of RMRP in the cartilage tissues (n = 10). *p < 0.05, **p < 0.01 and ***p < 0.001 B) Expression of FOCX1 and RBP4 in cartilage tissues was detected by western blot (n = 10). The OA cell models were constructed by treating chondrocytes derived from knee cartilage tissue of amputees without a history of OA with 5 μg/ml of LPS for 12 hours. C) CCK-8 assay was used to analyze cell viability (n = 3). D) RT-qPCR was employed to detect the expression of RMRP (n = 3). E) FOCX1 and RBP4 protein levels were checked by western blot (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001
RMRP knockdown inhibited LPS-induced chondrocyte apoptosis and inflammation
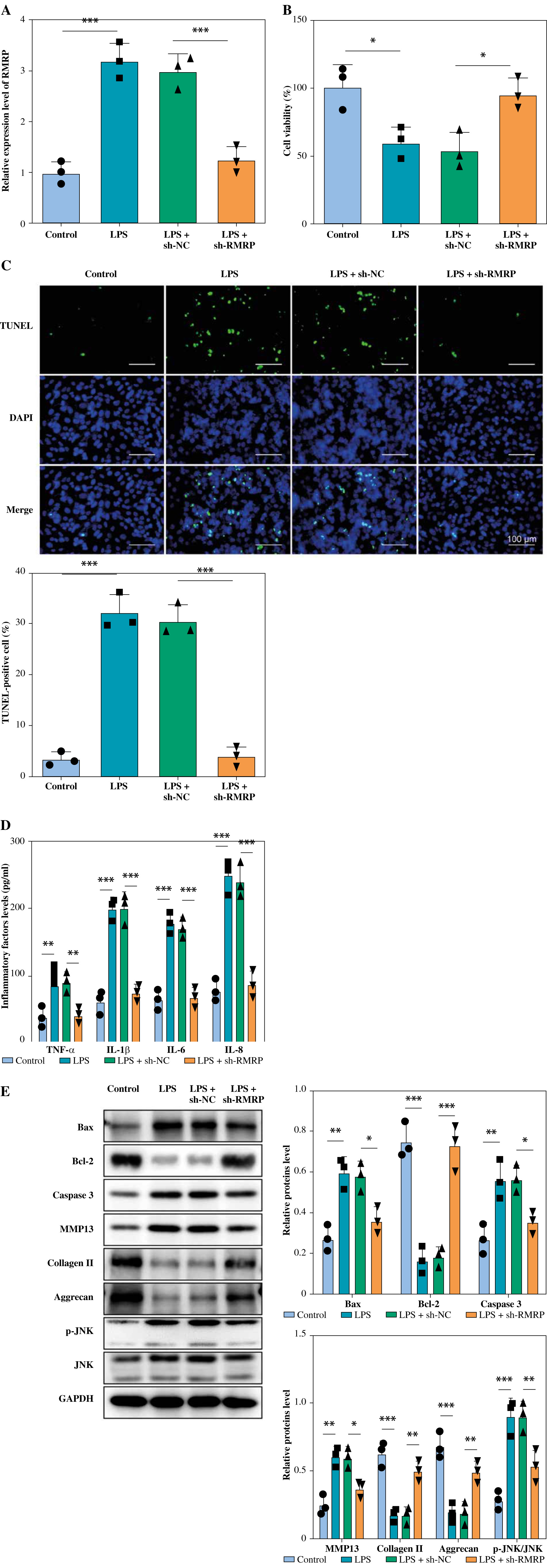
To further investigate the effect of RMRP in LPS-induced chondrocytes, chondrocytes were transfected with sh-RMRP and treated with LPS. The results showed that RMRP knockdown inhibited LPS-stimulated upregulation of RMRP in chondrocytes (Fig. 2A). In addition, LPS treatment significantly reduced chondrocyte viability and increased apoptosis; however, knockdown of RMRP reversed these results (Fig. 2B, C). Moreover, sh-RMRP could mitigate the elevated levels of pro-inflammatory factors (TNF-α, IL-1β, IL-6, and IL-8) induced by LPS stimulation (Fig. 2D). LPS treatment led to increased levels of Bax and caspase 3, but a decreased level of Bcl-2, which were eliminated by RMRP downregulation (Fig. 2E). Also, knockdown of RMRP relieved the LPS-induced elevation of MMP13 and reduction of collagen and aggrecan. Furthermore, LPS stimulation promoted JNK phosphorylation, but RMRP knockdown inhibited this result (Fig. 2E). Therefore, these results demonstrated that knockdown of RMRP could suppress LPS-induced chondrocyte apoptosis and inflammation.
Fig. 2
RMRP knockdown inhibited LPS-induced chondrocyte apoptosis and inflammation. sh-RMRP or sh-NC was transfected into chondrocytes isolated from cartilage tissues of amputees with no history of osteoarthritis (OA) and treated with 5 μg/ml LPS. A) RMRP levels were detected by RT-qPCR (n = 3). B, C) Cell viability and apoptosis were assessed by CCK-8 and TUNEL assays (scale bar = 100 μm) (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001. D) Levels of TNF-α, IL-1β, IL-6 and IL-8 were examined by ELISA kit (n = 3). E) Western blot was used to analyze the expression of Bax, Bcl-2, caspase 3, MMP13, collagen II, aggrecan, JNK and p-JNK (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001
RMRP bound with FOXC1 and promoted RBP4 expression
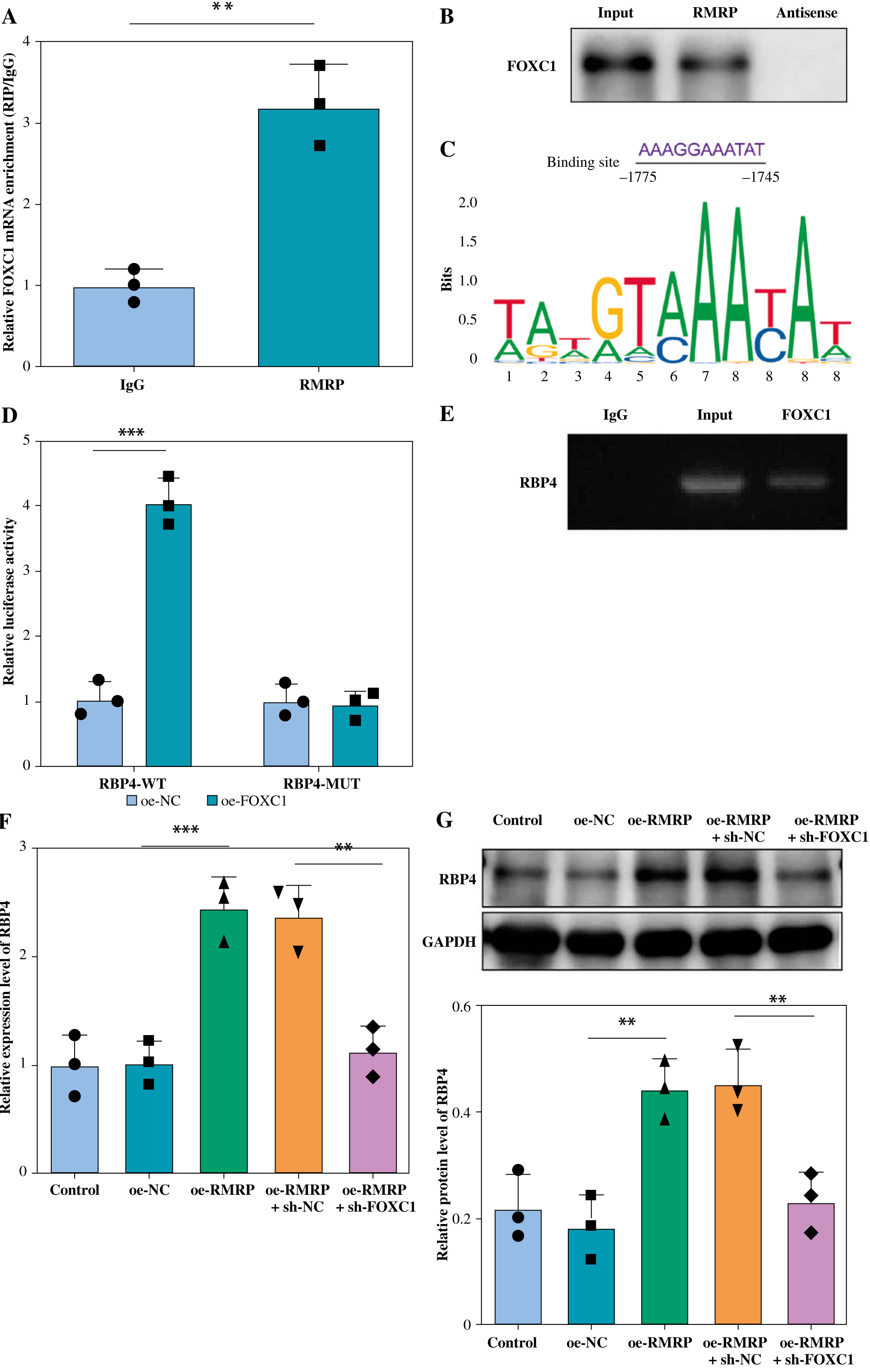
Next, we explored the downstream targets of RMRP in regulating OA chondrocyte apoptosis and inflammation. As shown in Figure 3A, it was confirmed that RMRP was directly bound with FOXC1, which was also demonstrated by RNA pull down (Fig. 3B). Using the JASPAR database, the current analysis suggested that FOXC1 had a possible binding site with RBP4 (Fig. 3C). Meanwhile, the results from dual-luciferase reporter assay subsequently demonstrated that FOXC1 overexpression significantly increased the luciferase activity presented by RBP4-WT but had no significant effect on that of RBP4-MUT (Fig. 3D), which was confirmed by ChIP assay (Fig. 3E). Overexpression of RMRP could upregulate the level of RBP4 in chondrocytes, but FOXC1 knockdown alleviated the promoting effect of RMRP overexpression on RBP4 (Fig. 3F, G), suggesting that FOXC1 transcriptionally activated RBP4. Overall, RMRP promoted the transcriptional activation of RBP4 by binding to FOXC1.
Fig. 3
RMRP bound to FOXC1 and promoted RBP4 expression. A, B) RIP and RNA pull-down assays were employed to verify the interaction between RMRP and FOXC1 (n = 3). C) JASPAR analyzed the potential binding site between FOXC1 and the RBP4 promoter region. D, E) The binding relationship between FOXC1 and RBP4 was examined by dual luciferase reporter and ChIP (n = 3). **p < 0.01 and ***p < 0.001. F, G) RT-qPCR and western blot were used to detect the RBP4 level (n = 3). **p < 0.01 and ***p < 0.001
FOXC1 upregulated RBP4 to promote apoptosis and inflammation in LPS-induced chondrocytes
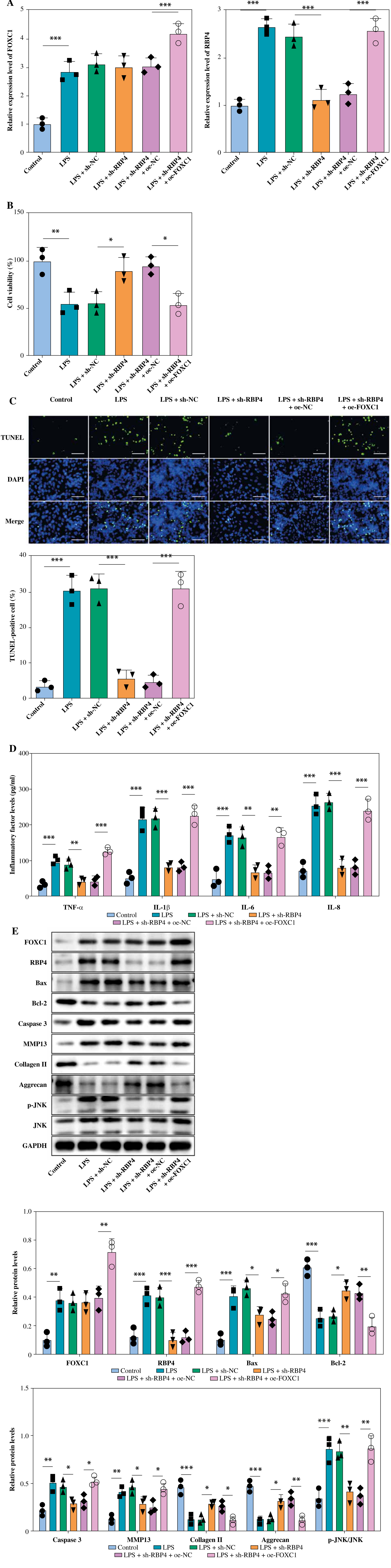
Chondrocytes were transfected with sh-RBP4 and oe-FOXC1 to investigate whether FOXC1 can promote LPS-induced chondrocyte apoptosis and inflammation through regulating RBP4. The RT-qPCR results showed that increased of FOXC1 and RBP4 levels were stimulated by LPS. However, RBP4 downregulation could inhibit RBP4 expression in chondrocytes under LPS treatment, while FOXC1 upregulation reversed the result. Also, FOXC1 overexpression further increased FOXC1 level in LPS-stimulated chondrocytes (Fig. 4A). Moreover, in LPS-induced chondrocytes, RBP4 knockdown mitigated the inhibitory effect of LPS on cell viability, which was overturned by FOXC1 upregulation (Fig. 4B). In contrast, the downregulation of RBP4 attenuated chondrocyte apoptosis in the presence of LPS and FOXC1 overexpression counteracted this anti-apoptotic effect of RBP4 (Fig. 4C). LPS treatment upregulated the levels of inflammatory cytokines, RBP4 knockdown significantly decreased the levels of TNF-α, IL-1β, IL-6 and IL-8 in LPS-induced chondrocytes, whereas FOXC1 overexpression counteracted this decrease (Fig. 4D). Additionally, RBP4 knockdown effectively prevented the upregulation of Bax, caspase 3, MMP13 and p-JNK, as well as the reduction of Bcl-2, collagen II, and aggrecan in LPS-induced chondrocytes, but FOXC1 overexpression reversed these effects (Fig. 4E). Consequently, FOXC1 activated RBP4 and promoted apoptosis and inflammation in LPS-induced chondrocytes.
Fig. 4
FOXC1 upregulated RBP4 to promote apoptosis and inflammation in LPS-induced chondrocytes. LPS-treated chondrocytes were transfected with sh-RBP4 and/or oe-FOXC1. A) Expression of RBP4 and FOXC1 was detected by RT-qPCR (n = 3). B) Cell viability was assessed using CCK-8 assay (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001. C) TUNEL assay was used to assess apoptosis (scale bar = 100 μm) (n = 3). D) Levels of TNF-α, IL-1β, IL-6, and IL-8 were detected using an ELISA kit (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001. E) Expression of FOXC1, RBP4, Bax, Bcl-2, caspase 3, MMP13, collagen II, aggrecan, JNK, and p-JNK was detected by western blot (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001. E) Expression of FOXC1, RBP4, Bax, Bcl-2, caspase 3, MMP13, collagen II, aggrecan, JNK, and p-JNK was detected by western blot (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001
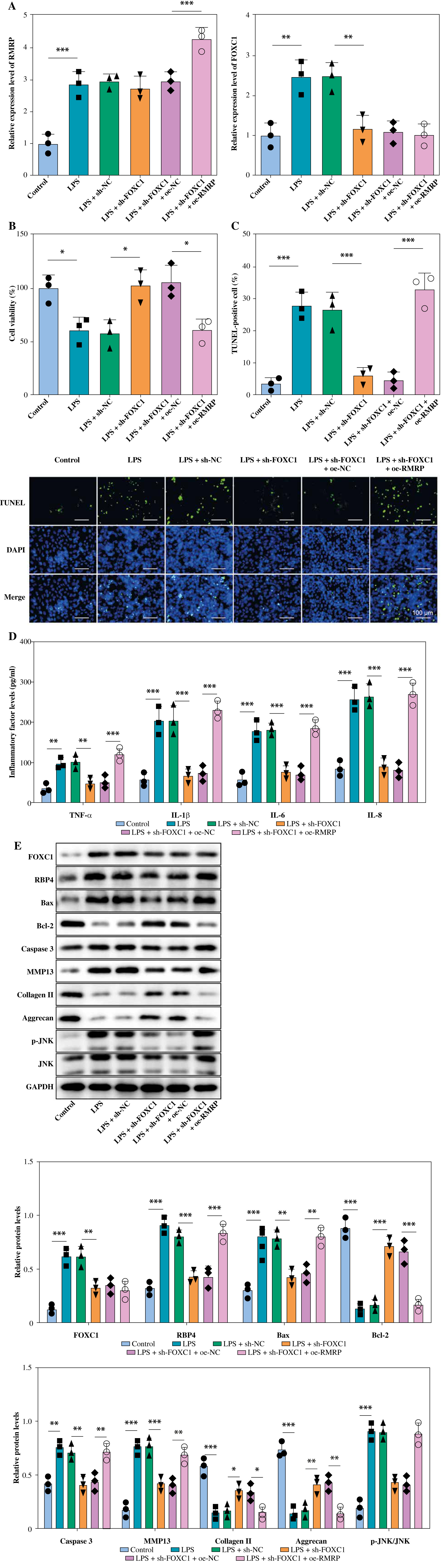
RMRP combined with FOXC1 to promote apoptosis and inflammation in LPS-induced chondrocytes
sh-FOXC1 and oe-RMRP were transfected into LPS-induced chondrocytes for subsequent studies. As shown in Figure 5A, LPS treatment increased the expression of RMRP and FOXC1 in chondrocytes. However, the knockdown of FOXC1 somewhat suppressed the LPS-induced upregulation of FOXC1, but it had no significant effect on RMRP, while overexpression of RMRP upregulated RMRP levels in chondrocytes exposed to LPS. Furthermore, FOXC1 knockdown not only ameliorated the LPS-induced decrease in cell viability but also protected against LPS-induced apoptosis, whereas overexpression of RMRP demonstrated the opposite effect (Fig. 5B, C). Additionally, a decrease in FOXC1 had a positive effect on reducing the levels of proinflammatory factors in chondrocytes exposed to LPS, while overexpression of RMRP did the opposite (Fig. 5D). Further, FOXC1 knockdown reduced the levels of FOXC1 and RBP4 in LPS-induced chondrocytes, while overexpression of RMRP alleviated the inhibition of RMRP by sh-FOXC1 (Fig. 5E). In chondrocytes exposed to LPS, FOXC1 knockdown effectively alleviated increases in Bax, caspase 3, MMP13 and p-JNK, while also counteracting decreases in Bcl-2, collagen II, and aggrecan. However, RMRP upregulation reversed the above results (Fig. 5E). To sum up, RMRP combined with FOXC1 and promoted apoptosis and inflammation in LPS-induced chondrocytes.
Fig. 5
RMRP combined with FOXC1 to promote apoptosis and inflammation of osteoarthritis (OA) chondrocytes. LPS-treated chondrocytes were transfected with sh-FOXC1 and oe-RMRP. A) RT-qPCR was used to determine the expression of RMRP and FOXC1 (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001. B) CCK-8 was used to detect cell viability (n = 3). C) Cell apoptosis was checked by TUNEL assay (scale bar = 100 μm) (n = 3). D) Levels of TNF-α, IL-1β, IL-6 and IL-8 were detected by ELISA kit (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001 E) Western blot was used to detect the expression of FOXC1, RBP4, Bax, Bcl-2, caspase 3, MMP13, collagen II, aggrecan, JNK and p-JNK (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001 E) Western blot was used to detect the expression of FOXC1, RBP4, Bax, Bcl-2, caspase 3, MMP13, collagen II, aggrecan, JNK and p-JNK (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001
Discussion
Osteoarthritis is a degenerative joint disease that seriously affects the quality of life of patients, causing a heavy burden on patients, families and society [30, 31]. It is generally believed that OA is a multifactorial disease, and its pathogenesis and pathologic processes need to be further investigated [32]. The main feature of OA is a loss of homeostatic balance between cartilage degradation and repair mechanisms due to diseased articular cartilage [33]. At present, the treatment of OA can only relieve pain and has no obvious effect in preventing or modifying OA disease [34]. The homeostasis of articular cartilage is determined by complex interactions between anabolism and catabolism, anti-inflammatory and pro-inflammatory effects, and anti-apoptotic mediators [35]. In this study, we used LPS to stimulate chondrocytes to construct a cellular model of OA cartilage injury. It was found that RMRP might be a key target for chondrocyte injury in OA. RMRP bound to FOXC1, upregulated FOXC1, and upregulated FOXC1 transcriptionally activated RBP4, which promoted the JNK pathway and ultimately triggered apoptosis and inflammation in LPS-induced chondrocytes.
Studies have shown that a large number of lncRNAs play a crucial role in OA. The lncRNA MALAT1 indirectly regulated AKT3 and inhibited chondrocyte apoptosis by acting as a sponge for miR-150-5p, thereby promoting OA development [11]. The lncRNA KLF3-AS1 promoted cartilage repair by promoting chondrocyte proliferation and inhibiting apoptosis through the miR-206/GIT1 axis [12]. Wang et al. observed that THUMPD3-AS1 enhanced the proliferation and inflammatory response of chondrocytes in OA [13]. In this study, we also discovered that lncRNA RMRP was significantly increased in both cartilage tissues of OA patients and LPS-induced chondrocytes. RMRP knockdown markedly increased the viability of chondrocytes, while significantly inhibiting apoptosis. Studies have revealed that proinflammatory factors are the key mediators of metabolic disruption processes in OA tissues [36]. In our study, knockdown of RMRP inhibited the expression of proinflammatory factors (TNF-α, IL-1β, IL-6, and IL-8). Articular cartilage is mainly composed of cartilage cells and ECM [35]. Extracellular matrix plays an important role in cartilage elastic support and biomechanical properties, of which collagen II and aggrecan are key components [1]. Pathological conditions such as inflammation can disrupt the microenvironment of cartilage ECM, leading to chondrocyte dysfunction and apoptosis, further exacerbating OA [37]. Our results demonstrated that knockdown of RMRP upregulated the levels of collagen II and aggrecan, and suppressed the expression of MMP13. In short, RMRP promoted chondrocyte apoptosis and inflammation in OA.
Next, we investigated the downstream factors of RMRP. In previous studies, FOXC1 has been identified as a key transcription factor involved in the development of OA [38]. In addition, a study found that miR-138-5p promoted IL-1β-induced degradation of the human cartilage ECM by targeting FOXC1 [21]. It is worth noting that we have proved the existence of a binding site between RMRP and FOXC1 through starBase prediction and experiments. Overexpression of RMRP promoted FOXC1 expression in chondrocytes of OA patients. At present, obesity is one of the important factors affecting the continuous increase in the incidence of OA [13]. Adipokines have been reported to play an essential role in cartilage homeostasis, metabolism, and inflammation [39]. A recent study revealed for the first time that the adipokine RBP4 was produced by cartilage and that it correlated with the levels of MMP1 and MMP3 in OA patients [27], but the specific molecular mechanism of RBP4 in OA was not analyzed. We found that FOXC1 transcription activated RBP4 in LPS-induced chondrocytes. Crucially, overexpression of RMRP upregulated the level of RBP4 while knocking down FOXC1 reversed this result. Therefore, we revealed for the first time that RMRP bound to FOXC1 and upregulated the expression of RBP4 in chondrocytes of OA patients.
We further elucidated the biological function of the regulatory network between RMRP, FOXC1 and RBP4 in chondrocytes of OA patients. This study found that knockdown of RBP4 and FOXC1 enhanced the viability of chondrocytes in OA, inhibited apoptosis and inflammation, and relieved the degradation of ECM. Notably, a study demonstrated that RBP4 could release proinflammatory factors (TNF-α and ILs) through the JNK pathway to promote inflammation [29]. In this study, RMRP combined with FOXC1 and promoted RBP4 transcriptional activation, which further activated the JNK signaling pathway, thereby promoting apoptosis and inflammation of chondrocytes in OA.
In summary, our research findings indicated that RMRP upregulated RBP4 by binding to FOXC1, further activating the JNK signaling pathway and promoting apoptosis and inflammation of chondrocytes in OA. Our findings provided new insights into the treatment of cartilage damage in OA. However, there is still a lack of animal experiments to further validate the role of lncRNA RMRP, FOXC1 and RBP4 regulatory networks in OA cartilage tissue injury, and more validation is necessary in the future.