
Introduction
Biliary atresia (BA) is a serious hepatobiliary disease in infants that is characterized by progressive intrahepatic and extrahepatic bile duct inflammation and fibrosis, resulting in bile stasis, progressive hepatic fibrosis, and cirrhosis [1]. In Asia, its incidence is approximately 100-500 cases per 100,000 infants in Taiwan and Japan [2]. Clinically speaking, BA is divided into three types: type I (approx. 5% of cases), in which obstruction occurs in the common bile duct and the gallbladder contains bile; type II (approx. 2% of cases), in which the obstruction site is the hepatic duct, the gallbladder does not contain bile but the proximal bile duct’s cavity contains bile; type III (> 90% of cases), with hilar bile duct obstruction and no bile in the proximal hepatic duct [3]. Currently, the most effective therapy for BA is Kasai portoenterostomy; however, there are still some patients, especially those with type III BA, whose problems are not solved by this procedure [4]. Hepatic fibrosis is a crucial feature of BA progression, and the activation of hepatic stellate cells (HSCs) is considered a major cause of liver fibrosis [5]. Therefore, suppressing hepatic fibrosis and HSC activation constitutes an approach to BA treatment.
Interleukin 17 (IL-17) is a cytokine implicated in the activation of a variety of immune cells and the inflammatory response [6]. Klemann et al. reported that IL-17 was apparently elevated in liver tissue samples obtained from patients with BA and IL-17 antibody inhibited liver inflammation and bilirubin levels to alleviate BA [7]. In the liver, CD4+ T cells can differentiate into helper T (Th)17 cells by producing IL-17 [8]. CD4+ T cells and the cytokines they produce can regulate HSC activation [9]. The IL-17 family includes IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F. A previous study found that IL-17A showed strong biological activity and was highest in Th17 cells [10]. The role of IL-17A in promoting liver fibrosis has been widely reported [9, 11]. Moreover, IL-17A has been reported to activate HSCs [12, 13]. Thus, this cytokine might affect HSC activation to promote hepatic fibrosis, thereby participating in BA progression. However, the mechanism of IL-17A in BA is still unclear. As previously described, IL-17A generally stabilizes the mRNA of downstream genes to mediate disease development [14, 15]. Moreover, some RNA-binding proteins (RBPs), including human antigen R (HuR), could interact with the adaptors tumor necrosis factor receptor (TNFR)-associated factor 2 (TRAF2) and TRAF5 to form a complex and regulate mRNA stability of downstream genes [16]. Here, we assumed that IL-17A mediated the mRNA stability of the downstream gene to promote BA progression by recruiting the TRAF2/TRAF5/HuR complex.
Glycolysis, the metabolic pathway that converts glucose to pyruvate, occurs in the cytoplasm and is induced by many metabolic enzymes [17]. Tian et al. proposed that D-2-hydroxyglutarate (D-2-HG) levels were abnormally elevated in patients with BA and promoted glycolysis to aggravate liver repair [18]. Glycolysis and HSC activation are closely related to liver fibrosis. For instance, it was reported that HSC activation was an initiating factor of hepatic fibrosis and it was accompanied by increasing glycolysis [19]. Thus, we thought that glycolysis probably promoted HSC activation and hepatic fibrosis in BA. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3), a major driver of glycolysis, is the most potent allosteric activator of phosphofructokinase-1 (PFK1, a rate-limiting enzyme in glycolysis) [20]. A previous study suggested that cytoplasmic polyadenylation element binding protein 4 (CPEB4)-induced PFKFB3 promoted glycolysis and HSC activation in mice and humans, eventually contributing to hepatic fibrosis [21]. The above information indicated that PFKFB3 might play a crucial role in BA.
Based on the information presented above, we put forward the hypothesis that IL-17A induces cell glycolysis by regulating the TRAF2/TRAF5/HuR signal axis, which further mediates PFKFB3 expression and stimulates HSC activation and fibrosis.
Material and methods
Collection of clinical samples
Referring to a previous study [22], we collected liver tissues from 16 patients with type III BA (BA group) and 14 patients with anicteric choledochal cysts (the non-BA group). Detailed information on our study participants is presented in Table 1. The diagnosis of BA was based on the residual fibrotic obstruction of the extrahepatic bile duct in the tissue removed after surgical cholangiography. The non-BA group was made up of patients with choledochal cysts without jaundice who had normal liver function and no history of immune-mediated disease. Liver fibro-mass tissues of the BA and non-BA groups were collected from the hepatic portal vein that was removed during Kasai’s surgery. After obtaining the samples, they were immediately frozen in liquid nitrogen for storage. Then, samples were maintained at –80°C before conducting experiments. This study was conducted according to the principles outlined in the Declaration of Helsinki (as revised in 2013). Our study was approved by the Ethics Committee of Hunan Children’s Hospital. All participants gave their written informed consent to participate in the study.
Table 1
Clinical characteristics in biliary atresia (BA) and anicteric choledochal cyst (non-BA) patients
[i] BA – biliary atresia, ALT – alanine aminotransferase, AST – aspartate aminotransferase, ALP – alkaline phosphatase, GGT – γ-glutamyl transpeptidase, DBIL – direct bilirubin, TBIL – total bilirubin. Anicteric choledochal cyst patients with nonicteric and normal liver function as controls (non-BA). Data are presented as mean ± standard deviation (SD)
Cell culture and treatment
LX-2 cells (BeNa, China), a line of human HSCs, express specific liver markers such as alpha-smooth muscle actin (α-SMA). These cells are star- or spindle-shaped. LX-2 cells were maintained in DMEM (Thermo Fisher Scientific, USA) supplemented with 10% FBS (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Beyotime, China) at 5% CO2 and 37°C. To investigate the role of IL-17A in BA, different concentrations (10, 30, and 100 ng/ml) of IL-17A (SRP3080, Merck, USA) were applied to treat LX-2 cells for 24 h. According to a previously published study [23], TGF-β1 (10 ng/ml, P01137, R&D Systems, USA), a substance that could activate HSCs, was used to treat LX-2 cells for 24 h. TGF-β1 served as the positive control for the IL-17A treatment group. 2-deoxy-D-glucose (2-DG; 5 mM, HY-13966, MedChemExpress, USA) was used as a glycolytic inhibitor.
Cell transfection
Short hairpin RNA targeting PFKFB3 (sh-PFKFB3), TRAF2 (sh-TRAF2), and TRAF5 (sh-TRAF5), and the HuR overexpression plasmid (oe-HuR) were obtained from GenePharma (China). Consistently, corresponding control groups including sh-NC and oe-NC were also obtained from GenePharma. LX-2 cells were seeded onto 6-well plates (2 × 105 cells/well) and allowed to grow until they were about 80% confluent. Then, the above plasmids or sequences were transfected into cells using Lipofectamine 3000 (Invitrogen, USA) according to the instructions. After 48 h, the transfection efficiency was confirmed via RT-qPCR or western blot. The transfected LX-2 cells were used for subsequent experiments.
RT-qPCR
Total RNA of clinical samples and LX-2 cells was extracted using the TRIzol reagent (R0011, Beyotime). Then, the purity and concentration of RNA were detected using the NanoDrop ND-1000 spectrophotometer (USA). When the ratio of the absorbance measured at 260 nm divided by the absorbance measured at 280 nm was greater than 1.9 and less than 2.1, the extracted RNA was considered to be of good purity and could be used for RT-qPCR. Afterward, cDNA synthesis was performed using the Script Reverse Transcription Reagent Kit (639505, TaKaRa, Japan). Quantitative real-time PCR was then conducted using SYBR Premix Ex Taq (RR820A, Takara) on an ABI Prism 7500 RT PCR system (Applied Biosystems, USA). The primer sequences are presented below.
IL-17A Forward: TCCCACGAAATCCAGGATGC
IL-17A Reverse: GGATGTTCAGGTTGACCATCAC
TRAF2 Forward: TCCCTGGAGTTGCTACAGC
TRAF2 Reverse: AGGCGGAGCACAGGTACTT
TRAF5 Forward: CCACTCGGTGCTTCACAAC
TRAF5 Reverse: GTACCGGCCCAGAATAACCT
PFKFB3 Forward: ATTGCGGTTTTCGATGCCAC
PFKFB3 Reverse: GCCACAACTGTAGGGTCGT
α-SMA Forward: ACTGAGCGTGGCTATTCCTCCGTT
α-SMA Reverse: GCAGTGGCCATCTCATTTTCA
Collagen I Forward: GTGCGATGACGTGATCTGTGA
Collagen I Reverse: CGGTGGTTTCTTGGTCGGT
HuR Forward: AACTACGTGACCGCGAAGG
HuR Reverse: CGCCCAAACCGAGAGAACA
β-actin Forward: TGGCACCACACCTTCTACAA
β-actin Reverse: CCAGAGGCGTACAGGGATAG
The PCR cycling conditions were set as follows: an initial denaturation at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s, annealing at 60°C for 30 s and extension at 72°C for 30 s. The relative expression of targeted genes was calculated using the formula 2-ΔΔCt. β-actin acted as the reference gene.
Western blot
RIPA lysis buffer (P0013B, Beyotime, China) containing phenylmethanesulfonyl fluoride (PMSF, ST2573, Beyotime) and phosphatase inhibitors (P1082, Beyotime) was used to acquire proteins from clinical samples (about 100 mg) and LX-2 cells (about 5 × 106 cells) with indicated treatments when the cell density was about 80%. The protein concentrations were then calculated using a BCA protein assay kit (P0009, Beyotime) through a standard curve obtained by preparing a series of standard concentrations. For the protein analysis, 30 µg of each protein sample was loaded in SDS-PAGE, which included 5% concentrated gel and 10% separation gel, after which it was isolated via SDS-PAGE electrophoresis. The isolated proteins were then transferred onto PVDF membranes at a voltage of 80 V for 1.5 h under ice bath conditions. The details of the primary antibodies are as follows: Antibodies against IL-17A (ab79056, 1 : 400), TRAF2 (ab126758, 1 : 1000), TRAF5 (ab303522, 1 : 1000), PFKFB3 (ab181861, 1 : 5000), α-SMA (ab5694, 1 : 100), collagen I (ab138492, 1 : 5000), Bax (ab32503, 1 : 5000), Bcl-2 (ab182858, 1 : 2000), solute carrier family 2 member 1 (GLUT1, ab115730, 1 : 100000), and HuR (ab200342, 1 : 1000) were purchased from Abcam (UK). Antibodies against hexokinase 2 (HK2, #2106, 1 : 1000) and β-actin (#4970, 1 : 1000) were obtained from CST (USA). Antibodies against PFK1 (MA5-47256, 1 : 1000), PKM2 (pyruvate kinase M1/2, PA5-29339, 1 : 8000) and lactate dehydrogenase A (LDHA, PA5-27406, 1 : 2000) were purchased from Thermo Fisher Scientific. β-actin was used as the internal parameter. After blocking with skimmed milk (5%), primary antibodies were provided for the incubation of PVDF membranes overnight at 4°C. Then, HRP-conjugated secondary antibody (A0208; 1 : 1000, Beyotime) was applied for membrane incubation for 1 h, the protein bands were observed using an ECL kit (P0018S; Beyotime) and the gray values were evaluated using Image J 5.0 (Bio-Rad, USA).
Detection of PFKFB3 mRNA stabilization
LX-2 cells with indicated treatments were seeded onto 6-well plates (2 × 105 cells/well). Then, 5 µg/ml of the transcriptional inhibitor known as actinomycin D (15021, CST, USA) was added to the cells to block messenger RNA (mRNA) transcription. At 0, 2, 4, 6, and 8 h after reaction, PFKFB3 mRNA expression was determined by RT-qPCR.
Cell Counting Kit-8 (CCK-8)
LX-2 cells were seeded onto 96-well plates (1 × 104 cells/well) and incubated overnight. Then, LX-2 cells were treated with 10, 30, 100 ng/ml IL-17A or 10 ng/ml TGF-β1 for 24 h. Afterward, 10 µl CCK-8 solution (C0038, Beyotime, China) was added to LX-2 cells in 96-well plates. After incubation for 2 h, the absorbance at 450 nm was examined by a microplate reader (Thermo Fisher Scientific).
5-Ethynyl-2'-deoxyuridine (EdU) incorporation assay
LX-2 cells were seeded on 96-well plates (1 × 104 cells/well) and incubated overnight. Afterward, LX-2 cells were treated with 50 µM EdU (C0078, Beyotime) for 2 h. LX-2 cells were then treated with 4% paraformaldehyde for 30 min and then were treated with 0.5% Triton X-100 for 10 min. Subsequently, LX-2 cells were stained with Hoechst (C1026, Beyotime) for 10 min. The stained cells were imaged under a fluorescent microscope (Olympus, Japan).
Detection of glucose uptake and lactate and ATP levels
LX-2 cells were seeded onto 96-well plates (1 × 104 cells/well) and incubated for 24 h. For the detection of glucose uptake, 2-NBDG, a fluorescent glucose uptake probe (Thermo Fisher Scientific) was used. After washing with Hanks’ Balanced Salt Solution (Thermo Fisher Scientific), the cells were incubated with 100 µM 2-NBDG for 30 min at 37°C. A microplate reader (Thermo Fisher Scientific) was used to detect the fluorescence value.
To evaluate lactate production, the lactate assay kit (1200011002; Eton Biosciences, USA) was used. The principle of this kit is that lactic acid is oxidized under the action of lactate dehydrogenase to produce a colored substance that can be detected at a wavelength of 450 nm. In short, LX-2 cells with indicated treatments were seeded onto 96-well plates (1 × 104 cells/well). According to the manufacturer’s instructions, firstly, a series of standard concentrations of lactate and samples were prepared and added along with other reagents corresponding to the kit. Afterward, the absorbances at 450 nm were determined. Finally, the lactate production of samples was calculated using a standard curve.
The ATP Assay Kit (S0026; Beyotime) was used for ATP level detection. This kit was developed according to the requirement for ATP energy when firefly luciferase catalyzes luciferin to produce fluorescence. In a certain concentration range, fluorescence production is proportional to the concentration of ATP. In brief, LX-2 cells with indicated treatments were implanted onto 6-well plates (1 × 106 cells/well) and cultured overnight. After lysis, the procedures were performed according to the manufacturer’s instructions. Finally, a microplate reader (Thermo Fisher Scientific) was applied to detect the fluorescence value.
Detection of extracellular acidification rate (ECAR)
Shortly, LX-2 cells were cultured in Seahorse XF 96 cell culture microplates at a density of 1 × 104 cells. The ECAR of LX-2 cells was measured using the XF96 Extracellular Flux analyzer (Seahorse Bioscience, USA). Following the procedure of ECAR detection, glucose, oligomycin, and 2-DG were added to microplates in an orderly manner. Of note, the ECAR values were recorded every 6 min. Data of ECAR were measured using the Seahorse XF24 software.
Co-immunoprecipitation (Co-IP) assay
LX-2 cells were lysed with the RIPA buffer (P0013B, Beyotime) on ice for 1 h. Subsequently, the lysate was centrifuged at 13,000 × g and 4°C for 10 min. Then, cell lysates were incubated overnight at 4°C with anti-HuR (ab200342; 1 : 30, Abcam) or IgG (ab172730; 1 : 5000, Abcam). To obtain the immunocomplexes, cell lysates were incubated on protein-G agarose beads (P2258; Beyotime) at 4°C for 4 h. After centrifugation, the agarose slurry was collected. The pellets were washed and resuspended in the SDS gel-loading buffer. Finally, TRAF2 and TRAF5 levels were detected by western blot analyses.
RNA immunoprecipitation (RIP)
A Magna RIP RNA-Binding Protein Immunoprecipitation Kit (17-700; Millipore, USA) was used to conduct the RIP assay. Around 1 × 107 LX-2 cells were harvested and lysed using the RIP lysis buffer for 5 min. Then, the anti- HuR (ab200342; 1 : 30, Abcam) or anti-IgG (ab172730; 1 : 5000, Abcam) antibodies were added to the solution including protein A/G agarose beads to prepare the anti- body-magnetic bead complex. After washing, the antibody-magnetic bead complex was added to the cell lysate at 4°C overnight. Finally, RNA enrichment pulled by the antibody-magnetic bead complex was examined using RT-qPCR.
RNA pull-down
Approximately LX2 cells (4 × 107) were collected and then lysed in 1 ml of the RIPA buffer. Cell lysates were subjected to centrifugation at 13,000 rpm for 10 min at 4°C. Biotinylated RNA probes against PFKFB3 were then incubated with cell lysates for 4 h at 37°C. Subsequently, the lysate was incubated with streptavidin-coated magnetic beads (88817; Invitrogen) at 4°C overnight. Then, after washing five times with the RIP wash buffer, the beads were collected. The HuR enrichment by biotinylated RNA probes against PFKFB3 was examined by western blot analyses.
Statistical analysis
All data are presented as the mean ± standard deviation (SD). For in vitro studies, three separate experiments were performed in triplicate. GraphPad Prism 9 was performed to analyze all data. Student’s t-test was performed to compare quantitative data between the two groups. The one-way analysis of variance (ANOVA) followed by the Tukey test was used for more than two groups of statistical analysis. The relationship between IL-17A and PFKFB3/TRAF2/TRAF5 was analyzed using Pearson’s correlation analysis. P < 0.05 was considered statistically significant.
Results
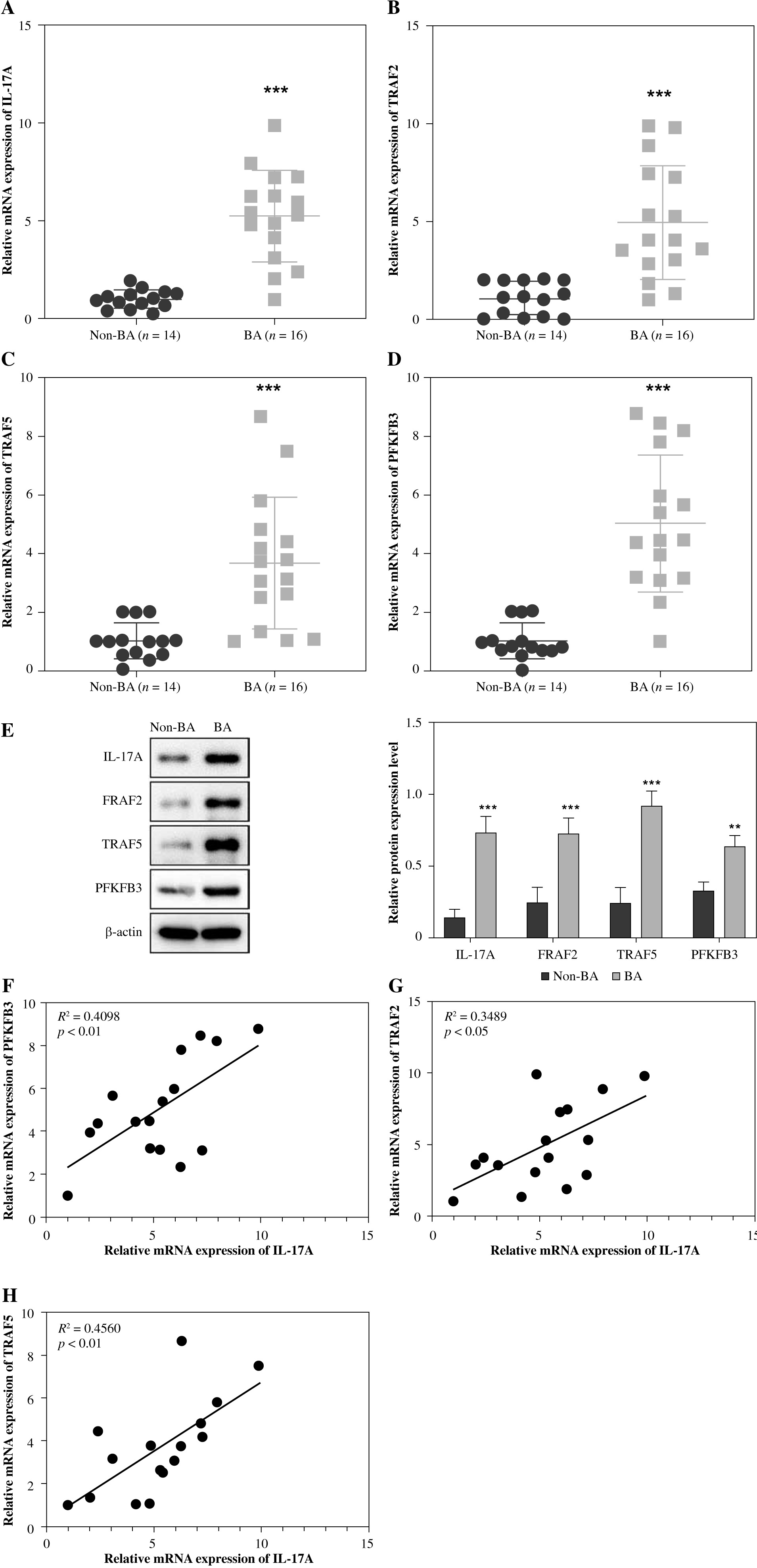
IL-17A, TRAF2, TRAF5, and PFKFB3 expression levels were abnormally elevated in liver tissue samples obtained from patients with BA
To investigate the abundance of IL-17A, TRAF2, TRAF5, and PFKFB3 in BA, liver tissue samples from 16 cases of BA and 14 cases of anicteric choledochal cyst (non-BA) were collected. We observed that the expression levels of IL-17A, TRAF2, TRAF5, and PFKFB3 were evidently elevated in BA groups compared with non-BA groups, which manifested at the RNA and protein levels (Fig. 1A-E). Pearson’s correlation analysis revealed that IL-I7A expression was positively correlated with PFKFB3 expression in BA (Fig. 1F). Moreover, a positive relationship between IL-17A expression and TRAF2/TRAF5 expression was also observed in BA (Fig. 1G, H). Taken together, molecules such as IL-17A, TRAF2, TRAF5, and PFKFB3 might be implicated in BA.
Fig. 1
Interleukin 17A, TRAF2, TRAF5, and PFKFB3 expression levels were abnormally elevated in the liver tissues of patients with biliary atresia (BA). Liver tissue samples from 16 patients with BA and 14 patients with an anicteric choledochal cyst without biliary atresia (non-BA) were collected. A-D) IL-17A, TRAF2, TRAF5, and PFKFB3 expression was detected using RT-qPCR (BA: n = 16; non-BA: n = 14). E) IL-17A, TRAF2, TRAF5, and PFKFB3 protein levels were measured by western blot analyses (n = 3, the experiments were conducted three times independently). **p < 0.01, ***p < 0.001; Student’s t-test (A-E). F) The correlation of expression level between IL-17A and PFKFB3 was evaluated by Pearson’s correlation analysis. G) The correlation of expression level between IL-17A and TRAF2 was evaluated by Pearson’s correlation analysis. H) The correlation of expression level between IL-17A and TRAF5 was evaluated by Pearson’s correlation analysis. **p < 0.01, ***p < 0.001; Pearson’s correlation analysis (F-H)
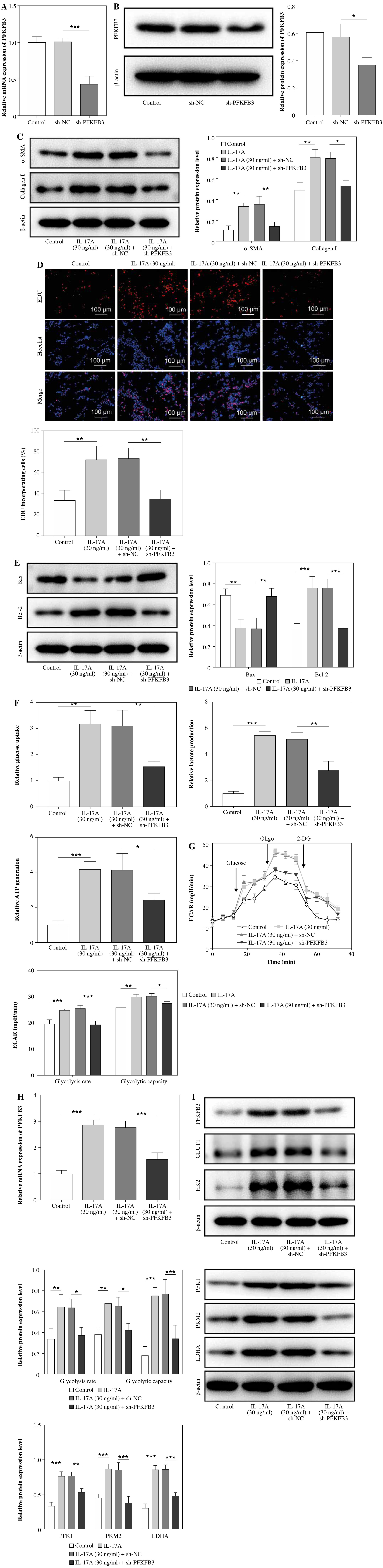
IL-17A promoted glycolysis to facilitate HSC activation and fibrosis
According to previous studies, HSC activation, liver fibrosis, and glycolysis are associated with BA progression [24, 25]. Therefore, we detected these three aspects after LX-2 cells were treated with IL-17A. As presented in Figure 2A, different concentrations of IL-17A (10, 30, and 100 ng/ml) increased the cell viability of LX-2 cells, of which 30 ng/ml of IL-17A had the most obvious promoting effect on cell viability, similarly to the effect of TGF-β1 (positive control), and when the concentration was upregulated to 100 ng/ml, the cell viability showed a slight downward trend compared with 30 ng/ml. In addition, the expression of fibrosis-related molecules (α-SMA and collagen I) was gradually elevated by IL-17A, with 30 ng/ml and 100 ng/ml IL-17A treatments, showing similar results to those of 10 ng/ml TGF-β1 (Fig. 2B, C). Based on these results, 30 ng/ml IL-17A was used for subsequent experiments. Tian et al. reported that β-amyloid could promote glycolysis to inhibit liver regeneration in BA, suggesting the aggravating effect of glycolysis on liver injury [26]. Then, IL-17A (30 ng/ml) and/or 2-DG (a glycolytic inhibitor) was added to LX-2 cells. We observed that IL-17A alone increased while 2-DG alone reduced α-SMA and collagen expression. Moreover, the combination of 2-DG and IL-17A impaired IL-17A-mediated upregulation of fibrosis-associated protein (α-SMA and collagen) expression (Fig. 2D). Additionally, IL-17A was elevated while 2-DG reduced LX2 cell proliferation, and the combination of 2-DG and IL-17A reversed IL-17A-induced LX2 cell proliferation (Fig. 2E). Furthermore, IL-17A increased Bcl-2 (an anti-apoptotic protein) and reduced Bax (a pro-apoptotic protein) expression, while 2-DG achieved the opposite effect. Of note, the combination of 2-DG and IL-17A compromised IL-17A-mediated alterations (Fig. 2F). As expected, IL-17A promoted glucose uptake and lactate production, and increased ATP levels, while 2-DG showed the opposite effect. Also, the combination of 2-DG and IL-17A eliminated these changes caused by IL-17A (Fig. 2G). Moreover, ECAR, used for assessing the metabolic status and energy status of cells, was investigated. ECAR analysis revealed that IL-17A treatment promoted glycolytic rate and glycolytic capability in LX-2 cells while 2-DG showed the opposite effect. Meanwhile, the combination of 2-DG and IL-17A could abolish IL-17A treatment-induced promoting effects (Fig. 2H). Subsequently, we observed that glycolytic-related proteins including PFKFB3, GLUT1, HK2, PFK1, PKM2, and LDHA were apparently elevated in BA patient liver tissues or IL-17A-treated LX-2 cells (Fig. S1). Additionally, PFKFB3, the key enzyme in glycolysis, was apparently elevated by IL-17A but reduced by 2-DG, and the IL-17A-induced elevation of PFKFB3 expression was reversed by the combination treatment of 2-DG and IL-17A (Fig. 2I). Western blot results revealed that glycolytic-related proteins (PFKFB3, GLUT1, HK2 PFK1, PKM2, and LDHA) were upregulated by IL-17A treatment and suppressed by 2-DG. The IL-17A-generated upregulation of these proteins was offset by the combination treatment of 2-DG and IL-17A (Fig. 2J). Collectively, IL-17A stimulated glycolysis to promote HSC activation, which might be involved in the upregulation of PFKFB3 expression.
Fig. 2
Interleukin 17A promoted glycolysis to facilitate HSC activation and fibrosis. LX-2 cells were treated with IL-17A (10, 30, and 100 ng/ml) or TGF-β1. A) Cell viability was examined using CCK-8. B, C) α-SMA and collagen I expression levels were determined by RT-qPCR and western blot analyses. LX-2 cells were treated with IL-17A (30 ng/ml) with/without 2-DG (a glycolytic inhibitor) D) α-SMA and collagen I expression levels were evaluated by western blot analyses. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-J. E) Cell proliferation was detected using the EdU assay. Scale bar: 100 μm. F) Bax and Bcl-2 expression levels were examined by western blot analyses. G) Glucose uptake, lactate production, and ATP levels were determined using commercial kits. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-J. H) ECAR was measured after sequential injections of glucose, oligomycin (Oligo), and 2-deoxyglucose (2-DG); ECAR acted as an indicator of glycolysis flux and glycolytic capacity. I) PFKFB3 expression was evaluated by RT-qPCR. J) PFKFB3, PFK1, PKM2, LDHA, GLUT1, and HK2 expression levels were investigated using western blot analyses. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-J

PFKFB3 silencing abolished IL-17A-induced glycolysis of HSCs
To investigate whether PFKFB3 participates in IL-17A-induced cell HSC activation and glycolysis, PFKFB3 expression was silenced in LX-2 cells with sh-PFKFB3 transfection (Fig. 3A, B). Then, LX-2 cells were treated with IL-17A, either alone or together with sh-PFKFB3 transfection. It was found that IL-17A treatment mediated the upregulation of α-SMA and collagen I was abolished in cells by PFKFB3 silencing (Fig. 3C). The IL-17A-mediated promotion of cell proliferation was compromised by PFKFB3 silencing (Fig. 3D). As expected, Bcl-2 expression was elevated and Bax expression was reduced by IL-17A treatment, while PFKFB3 silencing reversed these alterations (Fig. 3E). Additionally, PFKFB3 silencing reversed IL-17A-mediated upregulation of glucose uptake, lactate production and ATP levels (Fig. 3F). ECAR analysis also revealed that PFKFB3 silencing compromised IL-17A treatment-generated elevation of the glycolytic rate and glycolytic capability in LX-2 cells (Fig. 3G). Additionally, IL-17A treatment increased the levels of PFKFB3, PFK1, PKM2, LDHA, GLUT1, and HK2, while PFKFB3 knockdown attenuated these alterations caused by IL-17A (Fig. 3H, I). The abovementioned findings revealed that PFKFB3 knockdown attenuated IL-17A-induced cell proliferation and glycolysis in LX-2 cells.
Fig. 3
PFKFB3 silencing abolished IL-17-induced glycolysis in HSCs. A, B) PFKFB3 expression in LX-2 cells with sh-PFKFB3 transfection was measured using RT-qPCR and western blot analyses. LX-2 cells were subjected to IL-17 (30 ng/ml) treatment alone or together with sh-PFKFB3 transfection. C) α-SMA and collagen I expression levels were evaluated by western blot analyses. D) Cell proliferation was detected using the EdU assay. Scale bar: 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-I. E) Bax and Bcl-2 expression was examined by western blot analyses. F) Glucose uptake, lactate, and ATP levels were determined using commercial kits. G) ECAR was measured as an indicator of glycolysis flux and glycolytic capacity. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-I. H) PFKFB3 expression was evaluated by RT-qPCR. I) PFKFB3, PFK1, PKM2, LDHA, GLUT1, and HK2 expression was investigated using western blot analyses. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. Oneway analysis of variance (ANOVA) was employed for A-I
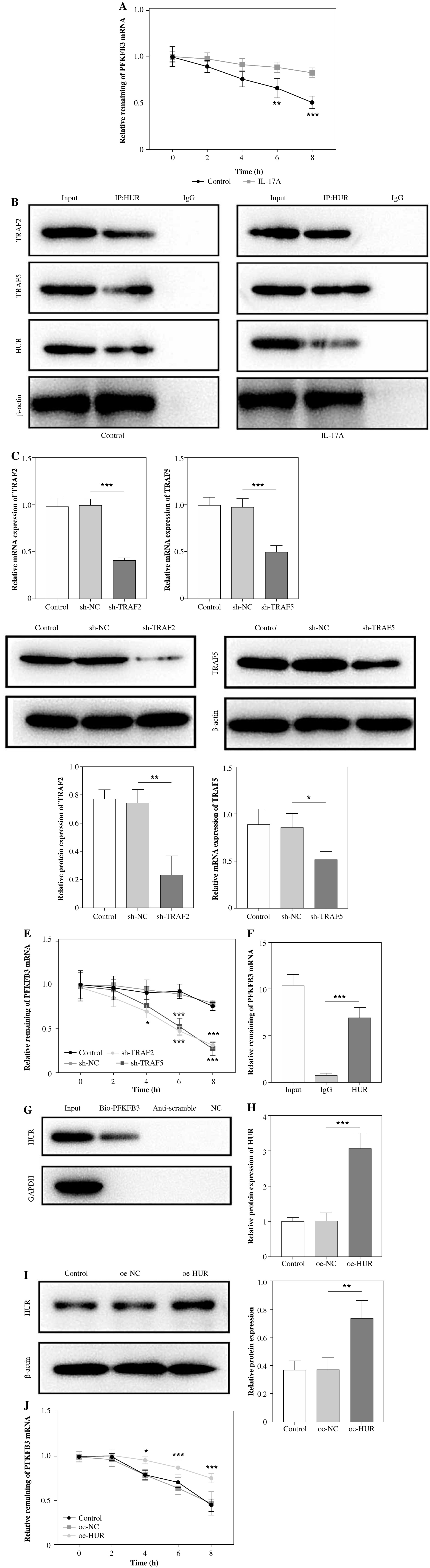
IL-17A maintained the stability of PFKFB3 mRNA by inducing formation of the TRAF2/TRAF5/HuR complex
To further explore why IL-17A increased the expression of PFKFB3, we treated LX-2 cells with the RNA synthesis inhibitor actinomycin D. The results indicated that IL-17A enhanced the mRNA stability of PFKFB3 (Fig. 4A). Amatya et al. proposed that IL-17A could recruit HuR (an RNA-binding protein) to the TRAF2/TRAF5 complex, thereby regulating the expression of downstream genes [3]. Herein, we used the Co-IP assay and found that the HuR antibody could enrich TRAF2 and TRAF5, and these enrichments were enhanced by IL-17A (Fig. 4B). The above indicated that IL-17A promoted formation of the TRAF2/TRAF5/HuR complex. Subsequently, we silenced TRAF2 or TRAF5 by transfecting sh-TRAF2 or sh-TRAF5 (Fig. 4C, D). After actinomycin D treatment, the mRNA stability of PFKFB3 was attenuated by the knockdown of TRAF2 or TRAF5 (Fig. 4E). Moreover, HuR was confirmed to interact with PFKFB3, as PFKFB3 could be enriched by the HuR antibody in the RIP assay and the HuR protein could be enriched by Bio-PFKFB3 in the RNA pull-down assay (Fig. 4F, G). Then, HuR expression was evidently elevated in LX-2 cells with oe-HuR transfection (Fig. 4H, I). As expected, HuR overexpression enhanced the mRNA stability of PFKFB3 (Fig. 4J). Taken together, these results showed that the IL-17A-mediated elevation of PFKFB3 was achieved via regulating formation of the TRAF2/TRAF5/HuR complex.
Fig. 4
Interleukin 17 maintained the stability of PFKFB3 mRNA by inducing formation of TRAF2/TRAF5/HuR complex. A) The stability of PFKFB3 mRNA was evaluated by RT-qPCR in IL-17-treated LX-2 cells upon actinomycin D treatment. B) The interactions among HuR, TRAF2, and TRAF5 were verified by Co-IP in LX-2 cells without/with IL-17A treatment. C) TRAF2 and TRAF5 expression levels were measured by RT-qPCR and western blot analyses in LX-2 cells with sh-TRAF2 or sh-TRAF5 transfection. *p < 0.05, **p < 0.01, ***p < 0.001. Student’s t-test for (A) and one-way analysis of variance (ANOVA) test for C-F. D) TRAF2 and TRAF5 expression levels were measured by RT-qPCR and western blot analyses in LX-2 cells with sh- TRAF2 or sh-TRAF5 transfection. E) The stability of PFKFB3 mRNA was evaluated by RT-qPCR in sh-TRAF2 or sh-TRAF5-transfected LX-2 cells upon actinomycin D treatment. F, G) The interaction between HuR and PFKFB3 was validated by RIP and RNA pull-down. H) HuR expression was determined using RT-qPCR and western blot analyses in LX-2 transfected with oe-HuR. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) test for C-F. I) HuR expression was determined using RT-qPCR and western blot analyses in LX-2 transfected with oe-HuR. J) The stability of PFKFB3 mRNA was evaluated by RT-qPCR in LX-2 cells with oe-HuR transfection upon actinomycin D treatment. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) test H-J
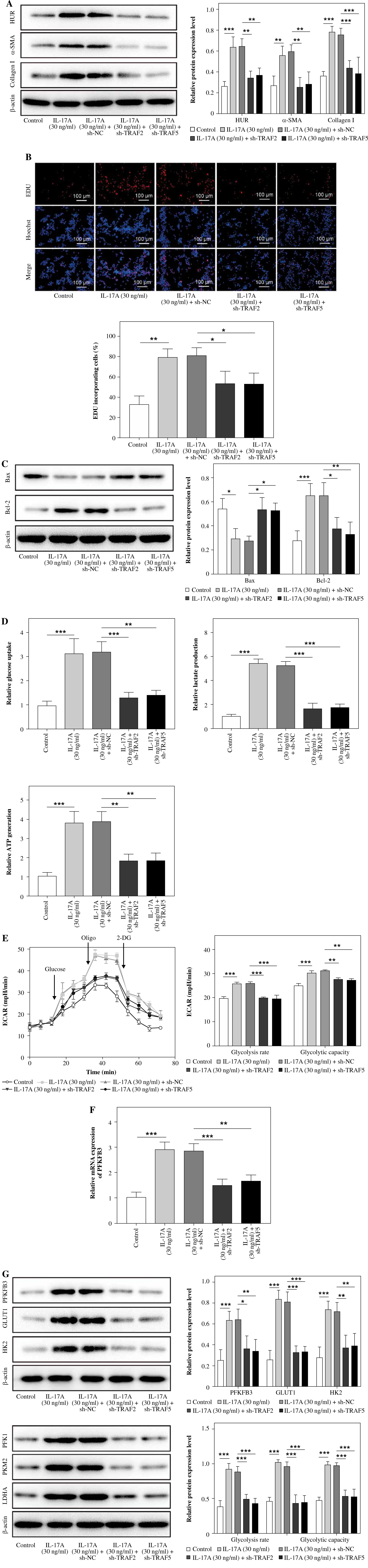
TRAF2 or TRAF5 knockdown impaired the IL-17A-induced glycolysis of HSCs
To investigate whether TRAF2 or TRAF5 is implicated in IL-17A-mediated promotion of cell proliferation and glycolysis in HSCs, sh-TRAF2 or sh-TRAF5 was transfected into LX-2 cells, following IL-17A treatment. As presented in Fig. 5A, IL-17A resulted in the increased expression of HuR, α-SMA, and collagen I, which was suppressed by TRAF2 or TRAF5 silencing. Also, IL-17A promoted cell proliferation and Bcl-2 expression and inhibited Bax expression in LX-2 cells; however, IL-17A-mediated influences were impaired by the silencing of TRAF2 or TRAF5 (Fig. 5B, C). For the influences of TRAF2 or TRAF5 silencing on IL-17A-induced glycolysis in LX-2 cells, TRAF2 or TRAF5 silencing compromised the IL-17A-mediated upregulation of glucose uptake, lactate production, and ATP levels (Fig. 5D). Moreover, IL-17A treatment increased the glycolysis rate and glycolysis capacity, and TRAF2 or TRAF5 knockdown reversed the promoting effects of IL-17A (Fig. 5E). Furthermore, the increase of PFKFB3 mRNA expression induced by IL-17A was weakened by the knockdown of TRAF2 or TRAF5 (Fig. 5F). IL-17A could greatly elevate PFKFB3, PFK1, PKM2, LDHA, GLUT1, and HK2 protein levels, while TRAF2 or TRAF5 knockdown inhibited IL-17A-generated alterations (Fig. 5G). In total, the IL-17A-induced glycolysis of HSCs was positively correlated with the expression of TRAF2 and TRAF5.
Fig. 5
TRAF2 or TRAF5 knockdown impaired IL-17A-induced glycolysis in HSCs. LX-2 cells underwent sh-TRAF2 or sh-TRAF5 transfection following IL-17A (30 ng/ml) treatment. A) HuR, α-SMA, and collagen I expression levels were determined by western blot analyses. B) Cell proliferation was detected using the EdU assay. Scale bar: 100 μm. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-G. C) Bax and Bcl-2 expression was examined by western blot analyses. D) Glucose uptake and lactate and ATP levels were determined using commercial kits. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-G. E) ECAR was measured as an indicator of glycolysis flux and glycolytic capacity. F) PFKFB3 expression was evaluated by RT-qPCR. G) PFKFB3, PFK1, PKM2, LDHA, GLUT1, and HK2 expression levels were investigated using western blot analyses. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-G. G) PFKFB3, PFK1, PKM2, LDHA, GLUT1, and HK2 expression levels were investigated using western blot analyses. n = 3; all experiments were conducted three times independently. *p < 0.05, **p < 0.01, ***p < 0.001. One-way analysis of variance (ANOVA) was employed for A-G
Discussion
Patients with BA, a devastating disease, have undergone surgery at an earlier stage to improve the cholestasis problem, yet the problem of hepatic fibrosis continues to progress further to cirrhosis [27]. Many studies have demonstrated that HSC activation contributes to liver fibrosis [28]. Thus, finding ways to inhibit HSC activation and liver fibrosis may provide an important way of alleviating BA. In this study, we found that IL-17A promoted glycolysis to enhance HSC activation and fibrosis by mediating the TRAF2/TRAF5/HuR/PFKFB3 axis.
As an inflammatory-related factor, IL-17 could be secreted by a variety of cells, such as T helper 17 (Th17) cells, CD4+/CD8+ T cells, γδ T cells, natural killer cells, innate lymphoid cells, and mast cells, among others [6]. IL-17 has been extensively investigated in multiple diseases, such as periodontitis, melanoma, and rheumatoid arthritis [29-31]. Several studies have reported that IL-17 was upregulated in patients with BA [7, 32], suggesting that IL-17 might have a vital effect on BA progression. In this study, we concentrated on IL-17A in BA. We found that IL-17A was highly expressed in liver tissue samples obtained from patients with BA, and IL-17A treatment significantly enhanced LX-2 cell viability and promoted HSC fibrosis. It has been reported that IL-17 could stabilize some mRNAs by recruiting RBPs (such as HuR, DDX3X, and Act1) through the TRAF2/TRAF5 adaptor [16, 33, 34]. For example, IL-17-induced Act1/TRAF2/TRAF5 complex formation to stabilize CXCL1 mRNA in pulmonary inflammation [35]. IL-17 promoted the mRNA stabilization of CXCL1 and CXCL5 by inducing the interactions of HuR with TRAF2/TRAF5 in inflammatory responses [16]. Bulek et al. proposed that TRAF2 and TRAF5 were essential adapter proteins in IL-17-mediated mRNA gene stabilization [35]. Here, we observed that the expression of TRAF2 and TRAF5 was increased in the hepatic tissues of patients with BA. In addition, we validated the interactions of HuR and TRAF2/TRAF5. We also found that IL-17A treatment promoted the interactions of HuR, TRAF2, and TRAF5. Our findings revealed that the TRAF2/TRAF5/HuR axis is important in IL-17A-mediated HSC activation.
HSC activation requires a lot of energy, and glycolysis serves as the main metabolic pathway of HSCs [36]. Growing evidence has revealed that glycolysis is associated with HSC activation and liver fibrosis [37]. For instance, costunolide inactivated HSCs by suppressing glycolysis in liver fibrosis [37]. Of note, two studies published in 2022 reported that glycolysis was positively correlated with BA progression [18, 26], suggesting that glycolysis is probably involved in BA. Thus, we introduced glycolysis to investigate how glycolysis affects IL-17A-mediated HSC activation and fibrosis. Our findings revealed that IL-17A promoted glycolysis in HSCs, while the glycolytic inhibitor 2-DG abolished IL-17A-mediated effects. Furthermore, 2-DG also reversed the IL-17A-mediated promotion of HSC activation and fibrosis, indicating that glycolysis was involved in IL-17A-mediated influences on HSC activation and liver fibrosis. PFKFB3, a well-known glycolytic trigger, was reported to be correlated with multi-fibrosis, including liver fibrosis [20, 21]. In the current study, abnormally higher PFKFB3 expression was observed in liver tissue samples obtained from patients with BA. Furthermore, PFKFB3 knockdown reversed IL-17A-induced glycolysis, HSC activation, and fibrosis. Subsequently, the reason for the IL-17-mediated elevation of PFKFB3 expression was investigated. IL-17 is known to promote TRAF2/TRAF5/HuR complex formation, which was similar to the results of this study. Herein, we found that IL-17A promoted PFKFB3 mRNA stabilization, which was reversed by TRAF2 or TRAF5 silencing.
In conclusion, the abovementioned information indicated that IL-17A promoted glycolysis by stabilizing PFKFB3 mRNA, which was associated with the IL-17A-induced formation of the TRAF2/TRAF5/HuR complex. Our results provided some experimental foundations for exploring the role of IL-17A in BA. Glycolysis is probably the key pathway for IL-17A to promote HSC activation and liver fibrosis.